
Lidé, mám dotaz:
Existuje způsob, jak získat 3,4-methylendioxyfenylpropan-2-on (MDP2P) z eugenolu?
Pokud ano, mohl by mi někdo poskytnout přesnou syntézu?
Našel jsem tuto syntézu, ale nechápu, jak mohu získat beta-nitroisoeugenol a β-Nitroisosafrol.
Problém je, že v mé zemi není možné sehnat sasafrasový olej, safrol, isosafrol a piperonal.
Do pětilitrové baňky se třemi hrdly vybavené míchadlem a zpětným chladičem bylo umístěno 1000 ml ethanolu a 110 g beta-nitroisoeugenolu. Směs se za stálého míchání zahřívala a po rozpuštění nitroisoeugenolu se přidalo 2500 ml horké vody. Za zahřívání a intenzivního míchání bylo přidáno 200 g redukovaného práškového železa a 8 g hydratovaného chloridu železitého. Za stálého míchání bylo pomalu přidáno 100 ml koncentrované kyseliny chlorovodíkové. Kyselina chlorovodíková způsobila prudkou reakci, která asi po 5 minutách ustoupila; směs byla za míchání refluxována po dobu dvou hodin a poté destilována za sníženého tlaku, dokud nebyly odebrány přibližně 2 litry destilátu. Zbytek byl přefiltrován a načechraný oxid železitý byl důkladně promyt horkou vodou a poté etherem. Spojený filtrát a výplachy byly silně okyseleny kyselinou chlorovodíkovou a extrahovány éterem. Eter byl vysušen a vydestilován za vzniku světlého oleje, který byl frakcionován ve vakuu za vzniku 68 g vanillylmethylketonu s teplotou varu 126-127 °C při 0,3 mm (výtěžek 72 %).
Z odpovídajících β-nitropropenylbenzenů byly téměř identickými postupy připraveny 3,4-dimethoxyfenylaceton a 3,4-methylenedioxyfenylaceton s výtěžky 90 a 72 %.
Příprava 2-MeO-P2P s toluenem/vodou jako rozpouštědlem viz Organic Syntheses, Coll. vol. 4, s. 573.
Moje modifikace této přípravy:
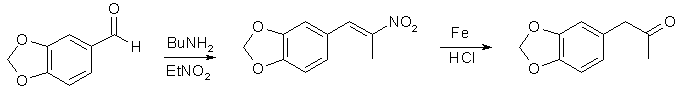
Do dvoulitrové baňky se třemi hrdly vybavené míchadlem a zpětným chladičem bylo umístěno 460 ml ethanolu1 a 67 g β-nitroisosafrolu2. Směs se za stálého míchání zahřívala a po rozpuštění žlutých krystalů se přidalo 1100 ml horké vody. Za zahřívání a intenzivního míchání bylo přidáno 80 g redukovaného práškového železa3 a 5 g FeCl3-6H2O. Za stálého míchání bylo za 30 min přidáno 63 ml koncetrované HCl. Směs byla za míchání 2 h refluxována a poté destilována za atmosférického tlaku4. Zbytek byl zfiltrován a hnědý oxid železitý byl extrahován 3x50 ml CH2Cl2. Filtrát a organické výplachy byly okyseleny HCl a byla sebrána červená ketonová vrstva. Světle zelená vrstva vody byla extrahována 2x100 ml CH2Cl2. Spojené extrakty byly vysušeny pomocí Na2SO4a vydestilovány za atmosférického tlaku (lázeň 80 °C). Dichlormethan byl zcela odstraněn ve vakuu za vzniku 55 g surového MDP2P jako tmavě červeného oleje, který byl použit pro reakci CH3NH2/Al.
Poznámky:
Existuje způsob, jak získat 3,4-methylendioxyfenylpropan-2-on (MDP2P) z eugenolu?
Pokud ano, mohl by mi někdo poskytnout přesnou syntézu?
Našel jsem tuto syntézu, ale nechápu, jak mohu získat beta-nitroisoeugenol a β-Nitroisosafrol.
Problém je, že v mé zemi není možné sehnat sasafrasový olej, safrol, isosafrol a piperonal.
Do pětilitrové baňky se třemi hrdly vybavené míchadlem a zpětným chladičem bylo umístěno 1000 ml ethanolu a 110 g beta-nitroisoeugenolu. Směs se za stálého míchání zahřívala a po rozpuštění nitroisoeugenolu se přidalo 2500 ml horké vody. Za zahřívání a intenzivního míchání bylo přidáno 200 g redukovaného práškového železa a 8 g hydratovaného chloridu železitého. Za stálého míchání bylo pomalu přidáno 100 ml koncentrované kyseliny chlorovodíkové. Kyselina chlorovodíková způsobila prudkou reakci, která asi po 5 minutách ustoupila; směs byla za míchání refluxována po dobu dvou hodin a poté destilována za sníženého tlaku, dokud nebyly odebrány přibližně 2 litry destilátu. Zbytek byl přefiltrován a načechraný oxid železitý byl důkladně promyt horkou vodou a poté etherem. Spojený filtrát a výplachy byly silně okyseleny kyselinou chlorovodíkovou a extrahovány éterem. Eter byl vysušen a vydestilován za vzniku světlého oleje, který byl frakcionován ve vakuu za vzniku 68 g vanillylmethylketonu s teplotou varu 126-127 °C při 0,3 mm (výtěžek 72 %).
Z odpovídajících β-nitropropenylbenzenů byly téměř identickými postupy připraveny 3,4-dimethoxyfenylaceton a 3,4-methylenedioxyfenylaceton s výtěžky 90 a 72 %.
Příprava 2-MeO-P2P s toluenem/vodou jako rozpouštědlem viz Organic Syntheses, Coll. vol. 4, s. 573.
Moje modifikace této přípravy:
Do dvoulitrové baňky se třemi hrdly vybavené míchadlem a zpětným chladičem bylo umístěno 460 ml ethanolu1 a 67 g β-nitroisosafrolu2. Směs se za stálého míchání zahřívala a po rozpuštění žlutých krystalů se přidalo 1100 ml horké vody. Za zahřívání a intenzivního míchání bylo přidáno 80 g redukovaného práškového železa3 a 5 g FeCl3-6H2O. Za stálého míchání bylo za 30 min přidáno 63 ml koncetrované HCl. Směs byla za míchání 2 h refluxována a poté destilována za atmosférického tlaku4. Zbytek byl zfiltrován a hnědý oxid železitý byl extrahován 3x50 ml CH2Cl2. Filtrát a organické výplachy byly okyseleny HCl a byla sebrána červená ketonová vrstva. Světle zelená vrstva vody byla extrahována 2x100 ml CH2Cl2. Spojené extrakty byly vysušeny pomocí Na2SO4a vydestilovány za atmosférického tlaku (lázeň 80 °C). Dichlormethan byl zcela odstraněn ve vakuu za vzniku 55 g surového MDP2P jako tmavě červeného oleje, který byl použit pro reakci CH3NH2/Al.
Poznámky:
- Použití methanolu místo ethanolu vedlo k neúplnému rozpuštění žlutých krystalů.
- β-Nitroisosafrol lze pohodlně připravit Knoevenagelovým postupem: 150 g piperonalu bylo smícháno se 76 ml nitroethanu (piperonal by se měl zcela rozpustit za vzniku bezbarvé kapaliny) a bylo přidáno 1,5 ml n-butylaminu(nebo n-pentylaminu). Vzniklý žlutý roztok byl uchováván na tmavém místě; po 2-3 dnech se začalo objevovat trochu vody, jak reakce probíhala. Světle oranžové krystaly produktu se začaly srážet po 6-8 dnech. Po 20 dnech byla směs zfiltrována. Světle hnědý filtrát byl po 5 dnech opět přefiltrován, čímž vznikl další produkt. Výtěžek 85-90 % světle oranžových krystalů, které jsou vhodné pro přípravu ketonu.
- Bylo použito železo redukované vodíkem. Použití práškového železa o velikosti 20 ok místo redukovaného železa vedlo k méně energické reakci, výsledný ethanol neobsahoval NH3 a keton byl kontaminován asi 20 % svého oximu (ne kvantitativní redukce).
- Byla odebrána frakce o teplotě 77-85 °C. Obsahovala určité množství NH3, které lze neutralizovat koncetrovanou H2SO4 a znovu vydestilovat přes Vigreuxovu kolonu. Výsledný 90% ethanol byl použit pro přípravu dalšího podílu ketonu.
