
Guys I have a question:
Is there a way to obtain 3,4-Methylenedioxyphenylpropan-2-one (MDP2P) from Eugenol?
If yes could someone give me a precise synthesis?
I found this synthesis but I dont understand how I can make beta-nitroisoeugenol and β-Nitroisosafrole.
The problem is that in my country it is impossible to get sassafras oil, safrole, isosafrole and piperonal.
In a 5-liter 3-necked flask equipped with a stirrer and reflux condenser were placed 1000 mL of ethanol and 110 g of beta-nitroisoeugenol. The mixture was heated with stirring and, when the nitroisoeugenol was dissolved, 2500 mL of hot water was added. With heating and vigorous stirring, 200 g of reduced iron powder and 8 g of hydrated ferric chloride were added. With continued stirring 100 mL of concentrated hydrochloric acid was added slowly. The hydrochloric acid caused a violent reaction which subsided after about 5 minutes; the mixture was refluxed with stirring for two hours and then distilled under reduced pressure until approximately 2 liters of distillate was collected. The residue was filtered, and the fluffy iron oxide was washed thoroughly with hot water and then with ether. The combined filtrate and washings were acidified strongly with hydrochloric acid and extracted with ether. The ether was dried and distilled to yield a light oil which was fractionated in vacuo to give 68 g of vanillyl methyl ketone boiling at 126-127°C at 0.3 mm (72% yield).
3,4-Dimethoxyphenylacetone and 3,4-methylenedioxyphenylacetone were prepared from the corresponding β-nitropropenylbenzenes by almost identical procedures with 90 and 72% yield.
For preparation of 2-MeO-P2P with toluene/water as solvent see Organic Syntheses, Coll. vol. 4, p. 573.
My modification of this preparation:
In a 2-liter 3-necked flask equipped with a stirrer and reflux condenser were placed 460 ml ofethanol1 and 67 g ofβ-nitroisosafrole2. The mixture was heated with stirring and, when the yellow crystals were dissolved, 1100 ml of hot water was added. With heating and vigorous stirring, 80 g of reduced ironpowder3 and 5 g FeCl3·6H2O were added. With continued stirring 63 ml conc. HCl was added in 30 min. The mixture was refluxed with stirring for 2 h and then distilled under atmosphericpressure4. The residue was filtered and brown iron oxide was extracted 3x50 ml CH2Cl2. Filtrate and organic washings were acidified with HCl and red ketone layer was collected. Light green water layer was extracted 2x100 ml CH2Cl2. The combined extracts were dried with Na2SO4and distilled at atmospheric pressure (80°C bath). Dichloromethane was completely removed in vacuo to give 55 g of crude MDP2P as deep red oil which was used for CH3NH2/Al reaction.
Notes:
Is there a way to obtain 3,4-Methylenedioxyphenylpropan-2-one (MDP2P) from Eugenol?
If yes could someone give me a precise synthesis?
I found this synthesis but I dont understand how I can make beta-nitroisoeugenol and β-Nitroisosafrole.
The problem is that in my country it is impossible to get sassafras oil, safrole, isosafrole and piperonal.
In a 5-liter 3-necked flask equipped with a stirrer and reflux condenser were placed 1000 mL of ethanol and 110 g of beta-nitroisoeugenol. The mixture was heated with stirring and, when the nitroisoeugenol was dissolved, 2500 mL of hot water was added. With heating and vigorous stirring, 200 g of reduced iron powder and 8 g of hydrated ferric chloride were added. With continued stirring 100 mL of concentrated hydrochloric acid was added slowly. The hydrochloric acid caused a violent reaction which subsided after about 5 minutes; the mixture was refluxed with stirring for two hours and then distilled under reduced pressure until approximately 2 liters of distillate was collected. The residue was filtered, and the fluffy iron oxide was washed thoroughly with hot water and then with ether. The combined filtrate and washings were acidified strongly with hydrochloric acid and extracted with ether. The ether was dried and distilled to yield a light oil which was fractionated in vacuo to give 68 g of vanillyl methyl ketone boiling at 126-127°C at 0.3 mm (72% yield).
3,4-Dimethoxyphenylacetone and 3,4-methylenedioxyphenylacetone were prepared from the corresponding β-nitropropenylbenzenes by almost identical procedures with 90 and 72% yield.
For preparation of 2-MeO-P2P with toluene/water as solvent see Organic Syntheses, Coll. vol. 4, p. 573.
My modification of this preparation:
In a 2-liter 3-necked flask equipped with a stirrer and reflux condenser were placed 460 ml ofethanol1 and 67 g ofβ-nitroisosafrole2. The mixture was heated with stirring and, when the yellow crystals were dissolved, 1100 ml of hot water was added. With heating and vigorous stirring, 80 g of reduced ironpowder3 and 5 g FeCl3·6H2O were added. With continued stirring 63 ml conc. HCl was added in 30 min. The mixture was refluxed with stirring for 2 h and then distilled under atmosphericpressure4. The residue was filtered and brown iron oxide was extracted 3x50 ml CH2Cl2. Filtrate and organic washings were acidified with HCl and red ketone layer was collected. Light green water layer was extracted 2x100 ml CH2Cl2. The combined extracts were dried with Na2SO4and distilled at atmospheric pressure (80°C bath). Dichloromethane was completely removed in vacuo to give 55 g of crude MDP2P as deep red oil which was used for CH3NH2/Al reaction.
Notes:
- Use of methanol instead of ethanol led to incomplete dissolution of yellow crystals.
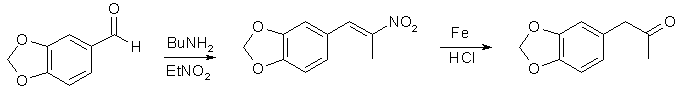
- β-Nitroisosafrole can be conveniently prepared by Knoevenagel: 150 g of piperonal was mixed with 76 ml nitroethane (piperonal should completely dissolve to give colourless liquid) and 1.5 ml of n-butylamine(or n-pentylamine) was added. Resulting yellow solution was held in a dark place; some water started appear after 2-3 days as the reaction proceeded. Light orange crystals of product started precipitate after 6-8 days. After 20 days a mixture was filtered. Light brown filtrate was filtered again after 5 days to afford next crop of product. Yield 85-90% of light orange crystals, which are suitable for preparation of the ketone.
- An iron reduced with hydrogen was used. Use of 20 mesh iron powder instead of reduced iron led to less vigorous reaction, resulting ethanol didn't contain NH3 and ketone was contamined with about 20% of its oxime (not quantitative reduction).
- A fraction 77-85°C was collected. It contained some NH3 which can be neutralized with conc. H2SO4 and distilled again via Vigreux column. Resulting 90% ethanol was used for preparation of the next portion of ketone.
