2C-B
4-BROMO-2,5-DIMETOXIFENETILAMINA
SÍNTESIS: Una solución de 100 g de 2,5-dimetoxibenzaldehído en 220 g de nitrometano se trató con 10 g de acetato de amonio anhidro y se calentó en baño de vapor durante 2,5 h con agitación ocasional. La mezcla de reacción, de color rojo intenso, se despojó del exceso de nitrometano al vacío y el residuo cristalizó espontáneamente. Este nitrostireno bruto se purificó moliendo bajo IPA, filtrando y secando al aire, para obtener 85 g de 2,5-dimetoxi-beta-nitrostireno como producto amarillo anaranjado de pureza adecuada para el paso siguiente. Puede purificarse aún más recristalizando a partir de IPA hirviendo.
En un matraz de fondo redondo de 2 L equipado con un agitador magnético y colocado bajo atmósfera inerte, se añadieron 750 mL de THF anhidro, conteniendo 30 g de LAH. A continuación se añadieron, en solución de THF, 60 g de 2,5-dimetoxi-beta-nitrostireno. La solución final era de color amarillo-marrón sucio, y se mantuvo a temperatura de reflujo durante 24 h. Después de enfriar, el exceso de hidruro se destruyó mediante la adición gota a gota de IPA. Después se añadieron 30 mL de NaOH al 15% para convertir los sólidos inorgánicos en una masa filtrable. La mezcla de reacción se filtró y la torta filtrada se lavó primero con THF y después con MeOH. Los licores madre combinados y los lavados se liberaron del disolvente al vacío y el residuo se suspendió en 1,5 L de H2O. Se acidificó con HCl, se lavó con 3x100 mL de CH2Cl2, se hizo fuertemente básico con NaOH al 25% y se volvió a extraer con 4x100 mL de CH2Cl2. Los extractos combinados se despojaron del disolvente al vacío, dando 26 g de residuo aceitoso, que se destiló a 120-130 °C a 0,5 mm/Hg para dar 21 g de un aceite blanco, 2,5-dimetoxi-fenetilamina
(2C-H) que capta dióxido de carbono del aire muy rápidamente.
A una disolución bien agitada de 24,8 g de 2,5-dimetoxifenetilamina en 40 mL de ácido acético glacial, se añadieron 22 g de bromo elemental disuelto en 40 mL de ácido acético. Tras un par de minutos, se produjo la formación de sólidos y la evolución simultánea de un calor considerable. Se dejó que la mezcla de reacción volviera a temperatura ambiente, se filtró y los sólidos se lavaron escasamente con ácido acético frío. Esta era la sal hidrobromuro. Hay muchas formas de sal complicadas, tanto polimorfos como hidratos, que pueden hacer que el aislamiento y la caracterización del 2C-B sean traicioneros. La ruta más feliz es formar la sal hidrobromuro insoluble mediante la base libre. Toda la masa de sal mojada en ácido acético se disolvió en H2O caliente, se hizo básica al menos a pH 11 con NaOH al 25% y se extrajo con 3x100 mL de CH2Cl2. La eliminación del disolvente dio 33,7 g de residuo que se destiló a 115-130 °C a 0,4 mm/Hg. El aceite blanco, 27,6 g, se disolvió en 50 mL de H2O que contenían 7,0 g de ácido acético. Esta solución clara se agitó enérgicamente y se trató con 20 mL de HCl concentrado. Se formó inmediatamente la sal anhidra de clorhidrato de 2,5-dimetoxi-4-bromofenetilamina (2C-B). Esta masa de cristales se eliminó por filtración (puede aflojarse considerablemente añadiendo otros 60 mL de H2O), se lavó con un poco de H2O y después con varias porciones de 50 mL de Et2O. Cuando se secó completamente al aire, se obtuvieron 31,05 g de finas agujas blancas, con una mp de 237-239 °C con descomposición. Cuando hay demasiado H2O presente en el momento de añadir el HCl concentrado final, se obtiene una forma hidratada de 2C-B. La sal de hidrobromuro funde a 214,5-215 °C. La sal de acetato tiene una mp de 208-209 °C.
DOSIFICACIÓN: 12 - 24 mg.
DURACIÓN: 4 - 8 h.
DOM
STP; 2,5-DIMETOXI-4-METILANFETAMINA
SÍNTESIS: A una solución de 54,9 g de 2,5-dimetoxi-4-metilbenzaldehído (véase la receta de
2C-D para su preparación) en 215 g de ácido acético glacial se añadieron 19,5 g de acetato de amonio anhidro y 30,6 g de nitroetano. Esta mezcla se calentó durante 3 h en el baño de vapor, la mezcla de reacción se enfrió en un baño de hielo húmedo, permitiendo la formación espontánea de cristales amarillos. Se añadió tanto H2O como fue posible (justo antes de que se formara un carácter aceitoso turbio persistente) y tras unas pocas h adicionales de reposo, el 1-(2,5-dimetoxi-4-metilfenil)-2-nitropropeno cristalino se retiró por filtración y se recristalizó a partir de ácido acético hirviendo. El rendimiento, tras secar hasta peso constante, fue de 28,3 g y el mp de 87-88 °C. Anal. (C12H15NO4) C, H ,N.
Una suspensión de 9,5 g de LAH en 750 mL de Et2O anhidro bien agitado se mantuvo a reflujo bajo atmósfera inerte, pasando el retorno del disolvente condensado a través de un cartucho Soxhlet que contenía 9,5 g de 1-(2,5-dimetoxi-4-metilfenil)-2-nitropropeno. Una vez completada la adición del nitrostireno, la suspensión agitada se mantuvo a reflujo durante 4 h más, después se enfrió a temperatura ambiente y se dejó seguir agitando durante toda la noche. El exceso de hidruro se destruyó mediante la adición de 750 mL de H2SO4 al 8%, con precaución, hasta que cesó la evolución del hidrógeno, y después a una velocidad que permitió la dispersión de los sólidos formados. Se separaron las fases, la fase acuosa se lavó una vez con Et2O, se trató con 225 g de tartrato sódico potásico y finalmente se hizo básica (pH >9) con NaOH al 5%. Se extrajo con 3x150 mL de CH2Cl2, se mezclaron los extractos y se eliminó el disolvente al vacío. El residuo fue 9,6 g de un aceite claro que formó espontáneamente cristales con una mp de 60,5-61 °C a partir de hexano. Estos sólidos se disolvieron en 150 mL de Et2O anhidro, y se saturaron con gas HCl anhidro. Tras reposar a temperatura ambiente durante 2 h, el clorhidrato cristalino de 2,5-dimetoxi-4-metilanfetamina (DOM) se extrajo por filtración, se lavó con Et2O y se secó al aire hasta peso constante. Se obtuvieron 8,25 g de cristales blancos brillantes que tenían una mp de 190,5-191,5 °C. El sulfato tuvo una mp de 131 °C. Anal. (C12H20ClNO2) C, H ,N.
El nitrostyrene antedicho se puede también convertir al producto final de la amina a través del intermediario del phenylacetone correspondiente. A una suspensión bien agitada de 10,4 g de hierro en polvo en 20 mL de ácido acético glacial mantenida a temperatura de reflujo, se añadieron 4,9 g de 1-(2,5-dimetoxi-4-metilfenil)-2-nitropropeno como sólido. Se continuó refluyendo durante 2 h y después se filtró todo a través de Celite húmedo. Después de lavar con 300 mL de H2O seguidos de 300 mL de Et2O, se separaron el filtrado combinado y los lavados, y la fase acuosa se extrajo con 2x100 mL de Et2O. La fase orgánica y los extractos se combinaron y se lavaron con 2x100 mL de K2CO3 saturado y el disolvente se eliminó al vacío dando un aceite rojizo que pesaba 3,3 g. Se destiló a 111-115 °C a 0,5 mm/Hg para dar un sólido verde pálido. Tras recristalización a partir de benceno, se obtuvieron 2,8 g de 1-(2,5-dimetoxi-4-metilfenil)-2-propanona en forma de cristales blancos con una mp de 57-59 °C. Esta cetona también se ha descrito como un aceite amarillo pálido con una pb de 115-118 °C a 0,4 mm/Hg. Una disolución de 0,7 g de 1-(2,5-dimetoxifenil-4-metil)-2-propanona en 20 mL de MeOH se trató con 6,0 g de acetato de amonio, 0,3 g de cianoborohidruro sódico y 3 g de tamices moleculares Linde 3 A. La mezcla se agitó durante una noche, y se disolvió en agua. La mezcla se agitó durante una noche, los sólidos se eliminaron por filtración y el filtrado se disolvió en 100 mL de H2O. La solución se acidificó con H2SO4 diluido y se lavó con 2x25 mL de CH2Cl2. La fase acuosa se hizo básica con NaOH acuoso, y el producto se extrajo con 2x25 mL de CH2Cl2. El disolvente se eliminó al vacío y el residuo se destiló (a 160 °C a 0,2 mm/Hg) para dar un producto incoloro que se disolvió en 3 mL de IPA, se neutralizó con HCl concentrado y se diluyó con 50 mL de Et2O anhidro. Se obtuvieron 0,18 g de clorhidrato de 2,5-dimetoxi-4-metilanfetamina (DOM) como un sólido blanco con una mp de 187-188 °C.
Los isómeros ópticos de DOM se han preparado de dos formas. La base racémica se ha resuelto como la sal de ácido orto-nitrotartranílico por recristalización de EtOH. El ácido (+) proporciona preferentemente el isómero (+) o "S" de la DOM. Además, la mencionada 1-(2,5-dimetoxi-4-metilfenil)-2-propanona puede aminarse reductivamente con alfa-metil-bencilamina ópticamente activa con Níquel Raney. Esta amina se aísla y purifica por recristalización de la sal clorhidrato. Cuando era ópticamente pura, el grupo bencilo se eliminó por hidrogenólisis con paladio sobre carbono. La mp de cualquiera de los isómeros ópticos, como sales de clorhidrato, fue de 204-205 °C.
DOSIFICACIÓN: 3 - 10 mg.
DURACIÓN: 14 - 20 h.
MDA
3,4-METILENDIOXIANFETAMINA
SÍNTESIS: (a partir de piperonal) A una solución de 15,0 g de piperonal en 80 mL de ácido acético glacial se añadieron 15 mL de nitroetano seguidos de 10 g de ciclohexilamina. La mezcla se mantuvo a temperatura de baño de vapor durante 6 h, se diluyó con 10 mL de H2O, se sembró con un cristal de producto y se enfrió toda la noche a 10 °C. Los cristales amarillos brillantes se eliminaron por filtración y se secaron al aire para obtener 10,7 g de 1-(3,4-metilendioxifenil)-2-nitropropeno con una mp de 93-94 °C. Se elevó a 97-98 °C. Ésta se elevó a 97-98 °C por recristalización a partir de ácido acético. Los esfuerzos más convencionales de síntesis del nitrostireno utilizando un exceso de nitroetano como disolvente y acetato de amonio anhidro como base, dan un producto impuro en rendimientos muy pobres. El nitrostireno se ha obtenido con éxito a partir de los componentes en MeOH frío, con NaOH acuoso como base.
Se colocó una suspensión de 20 g de LAH en 250 mL de THF anhidro bajo atmósfera inerte y se agitó magnéticamente. Se añadieron, gota a gota, 18 g de 1-(3,4-metilendioxifenil)-2-nitropropeno en disolución en THF y la mezcla de reacción se mantuvo a reflujo durante 36 h. Después de llevarla de nuevo a temperatura ambiente, el exceso de hidruro se destruyó con 15 mL de IPA, seguido de 15 mL de NaOH al 15%. Se añadieron 50 mL adicionales de H2O para completar la conversión de las sales de aluminio en un sólido suelto, blanco y fácil de filtrar. Se eliminó por filtración y la torta filtrada se lavó con THF adicional. El filtrado y los lavados combinados se eliminaron del disolvente al vacío y el residuo se disolvió en H2SO4 diluido. El lavado con 3x75 mL de CH2Cl2 eliminó gran parte del color, y la fase acuosa se hizo básica y se volvió a extraer con 3x100 mL de CH2Cl2. Al eliminar el disolvente se obtuvieron 13,0 g de un aceite de color amarillo que se destiló. La fracción que hirvió a 80-90 °C a 0,2 mm pesó 10,2 g y era blanca como el agua. Se disolvió en 60 mL de IPA, se neutralizó con HCl concentrado y se diluyó con 120 mL de Et2O anhidro que produjo una turbidez duradera. Se formaron cristales espontáneamente que se eliminaron por filtración, se lavaron con Et2O y se secaron al aire para proporcionar 10,4 g de clorhidrato de 3,4-metilendioxianfetamina (MDA) con una mp de 187-188 °C.
(a partir de 3,4-metilendioxifenilacetona) A una solución de 32,5 g de acetato de amonio anhidro en 120 mL de MeOH, se añadieron 7,12 g de 3,4-metilendioxifenilacetona (véase MDMA para su preparación) seguidos de 2,0 g de cianoborohidruro sódico. La solución amarilla resultante se agitó enérgicamente y se añadió periódicamente HCl concentrado para mantener el pH de la mezcla de reacción entre 6 y 7, según se determinó con papel de pH universal externo húmedo. Después de varios días, quedaron sólidos sin disolver en la mezcla de reacción y no se necesitó más ácido. La mezcla de reacción se añadió a 600 mL de HCl diluido y se lavó con 3x100 mL de CH2Cl2. Los lavados combinados se volvieron a extraer con una pequeña cantidad de HCl diluido, se combinaron las fases acuosas y se volvieron básicas con NaOH al 25%. A continuación se extrajo con 3x100 mL de CH2Cl2, se combinaron estos extractos y se eliminó el disolvente al vacío para obtener 3,8 g de un residuo de color rojo. Se destiló a 80-90 °C a 0,2 mm/Hg para obtener 2,2 g de un aceite absolutamente blanco como el agua. No hubo formación obvia de una sal de carbonato cuando se expuso al aire. Se disolvió en 15 mL de IPA, se neutralizó con 25 gotas de HCl concentrado y se diluyó con 30 mL de Et2O anhidro. Lentamente se produjo la deposición de cristales blancos de clorhidrato de 3,4-metilendioxianfetamina (MDA) que pesó 2,2 g y tuvo una mp de 187-188 °C. La preparación de la formamida (un precursor de la MDMA) y la acetamida (un precursor de la MDE) se describen en esas entradas.
DOSIFICACIÓN: 80 - 160 mg.
DURACIÓN: 4 - 6
(revisado, Sep 2001).
MESCALINA;
3,4,5-TRIMETOXIFENETILAMINA
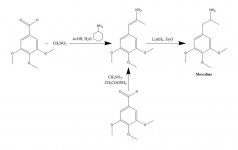
SÍNTESIS: Una disolución de 20 g de 3,4,5-trimetoxibenzaldehído, 40 mL de nitrometano y 20 mL de ciclohexilamina en 200 mL de ácido acético se calentó en baño de vapor durante 1 h. A continuación, la mezcla de reacción se diluyó lentamente y con buena agitación, con 400 mL de H2O, lo que permitió la formación de una pesada masa cristalina amarilla. Ésta se eliminó por filtración, se lavó con H2O y se aspiró lo más seca posible. La recristalización a partir de MeOH hirviendo (15 mL/g) dio, tras filtración y secado al aire, beta-nitro-3,4,5-trimetoxiestireno como cristales amarillos brillantes que pesaban 18,5 g. Una síntesis alternativa fue eficaz, utilizando un exceso de nitrometano como disolvente y como reactivo, si la cantidad de acetato de amonio catalizado se mantenía pequeña. Una solución de 20 g de 3,4,5-trimetoxibenzaldehído en 40 mL de nitrometano que contenía 1 g de acetato de amonio anhidro se calentó en baño de vapor durante 4 h. El disolvente se extrajo al vacío y el aceite amarillo residual se disolvió en dos volúmenes de MeOH caliente, se decantó de algunos insolubles y se dejó enfriar. Los cristales formados se eliminaron por filtración, se lavaron con MeOH y se secaron al aire dando 14,2 g. de cristales amarillos brillantes de beta-nitro-3,4,5-trimetoxiestireno. El uso de estas proporciones pero con 3,5 g de acetato de amonio dio amplios productos de reacción secundaria incluso cuando se trabajó después de sólo 1,5 h de calentamiento. El rendimiento de nitrostireno fue, en este último caso, insatisfactorio.
A una suspensión a reflujo suave de 2 g de LAH en 200 mL de Et2O, se añadieron 2,4 g de beta-nitro-3,4,5-trimetoxiestireno como solución saturada de Et2O utilizando un condensador de extracción Soxhlet modificado para permitir el retorno continuo del disolvente condensado a través del cartucho. Una vez completada la adición, se mantuvieron las condiciones de reflujo durante otras 48 h. Tras enfriar la mezcla de reacción, se añadió con precaución un total de 150 mL de H2SO4 1,5 N, destruyendo el exceso de hidruro y proporcionando finalmente dos fases claras. Se separaron y la fase acuosa se lavó una vez con 50 mL de Et2O. A continuación se añadieron 50 g de tartrato sódico potásico, seguidos de NaOH suficiente para que el pH fuera >9. Se extrajo con 3x3 mL de Et2O. A continuación se extrajo con 3x75 mL de CH2Cl2, y el disolvente de los extractos agrupados se eliminó al vacío. El residuo se destiló a 120-130 °C a 0,3 mm/Hg dando un aceite blanco que se disolvió en 10 mL de IPA y se neutralizó con HCl concentrado. Los cristales blancos que se formaron se diluyeron con 25 mL de Et2O, se eliminaron por filtración y se secaron al aire para proporcionar 2,1 g de clorhidrato de 3,4,5-trimetoxifenetilamina (M) como cristales blancos brillantes. La sal de sulfato formaba cristales espectaculares a partir del agua, pero tenía un mp amplio y poco característico. Una síntesis alternativa puede emplear 3,4,5-trimetoxifenilacetonitrilo, como se describe en beta-D.
DOSIFICACIÓN: 200-400 mg (como sal sulfato), 178-356 mg (como sal clorhidrato)
[Nota de Erowid: El texto original decía "178-256" pero era un error. El error fue encontrado por Bo y verificado con Shulgin. Ver la página de Dosis de Mescalina de Erowid
para una discusión más completa de las formas de mescalina].
DURACIÓN: 10-12 h
TMA
3,4,5-TRIMETOXIANFETAMINA
SÍNTESIS: A una disolución de 39,2 g de 3,4,5-trimetoxibenzaldehído en 30 mL de EtOH caliente se añadieron 15,7 g de nitroetano seguidos de 1,5 mL de n-butilamina. La mezcla de reacción se dejó reposar a 40 °C durante 7 días. Enfriando y rascando se obtuvieron finas agujas amarillas que, tras eliminarlas por filtración y secado al aire, pesaron 48 g. La recristalización a partir de EtOH dio 2-nitro-1-(3,4,5-trimetoxifenil)propeno como cristales amarillos con una mp de 94-95 °C. Anal. (C12H15NO5) C,H,N. Alternativamente, una disolución de 20 g del aldehído en 75 mL de nitroetano se trató con 4 g de acetato amónico anhidro y se calentó en baño de vapor hasta que se generó un color rojo intenso. La eliminación del exceso de disolvente/reactivo al vacío dio un aceite rojo que se disolvió en un volumen igual de MeOH hirviendo. Al enfriar, se separaron cristales amarillos del nitropropeno. La recristalización a partir de MeOH dio, tras secado al aire hasta peso constante, 13,0 g con el mismo mp.
Bajo atmósfera inerte, se mojaron 38 g de LAH con 100 mL de Et2O anhidro y se suspendieron en 1 L de THF seco. Se llevó a reflujo suave y se añadió lentamente una solución de 43,7 g de 2-nitro-1-(3,4,5-trimetoxifenil)propeno en 160 mL de THF. Se continuó refluyendo durante 36 h y después se enfrió la mezcla de reacción con un baño de hielo externo. El exceso de hidruro se destruyó mediante la adición prudente de 38 mL de H2O, a lo que siguieron 38 mL de NaOH al 15% y, finalmente, otros 114 mL de H2O. Las sales inorgánicas, que deberían haber acabado como una masa suelta, granulada y fácil de filtrar, parecían más bien pasta de biblioteca, pero se filtraron de todos modos. Se intentó lavar con THF, pero no resultó eficaz. El filtrado y los lavados combinados se eliminaron del disolvente al vacío, obteniéndose 31,5 g de la base bruta como aceite ámbar. Se disolvió en 140 mL de IPA, se neutralizó con HCl concentrado (se necesitaron 15 mL) y se diluyó con 650 mL de Et2O anhidro. Hubo una fase oleosa inicial que al continuar agitando cambió a sólidos rosa pálido. Éstos se molieron finamente bajo CH3CN para dar 15,2 g de clorhidrato de 3,4,5-trimetoxianfetamina (TMA) como cristales blancos que fundieron a 195-211 °C. Todas las sales de aluminio de todas partes se disolvieron en HCl diluido, y se añadió 1 Kg de tartrato sódico potásico. A continuación se añadió NaOH al 25% para llevar el pH a >9 sin que se produjera la precipitación de alúmina básica. La extracción de esta fase con CH2Cl2 fue seguida por la eliminación del disolvente y la formación de sales como se ha descrito anteriormente, permitió el aislamiento de 6,4 g adicionales de TMA. El producto así preparado contiene entre un 10 y un 15% de 3,5-dimetoxi-4-hidroxianfetamina como impureza. Una solución de 20 g de TMA preparada de esta manera en 200 mL de NaOH al 5% se extrajo con 2x200 mL de CH2Cl2. Los extractos combinados se lavaron con 4x100 mL de NaOH al 5%, y los lavados acuosos se combinaron con la fase base original. La fase orgánica se despojó de su CH2Cl2 al vacío para dar un aceite que se disolvió en 40 mL de IPA, se neutralizó con HCl concentrado y se diluyó con 400 mL de Et2O anhidro. Inmediatamente se formaron espectaculares cristales blancos de clorhidrato puro de 3,4,5-trimetoxianfetamina, con un peso de 15,4 g y una mp de 220-221 °C. La fase acuosa se llevó a neutralidad, se trató con 10 g de dihidrógeno fosfato potásico, se llevó a pH 9,0 con la adición cuidadosa de NaOH y se extrajo con 5x100 mL de CH2Cl2. La evaporación del disolvente al vacío dio un aceite que cristalizó espontáneamente. Este producto, 3,5-dimetoxi-4-hidroxianfetamina, pudo purificarse más por
sublimación a 130 °C a 0,2 mm/Hg. Era un sólido cristalino blanco que se decoloraba lentamente en el aire. La bibliografía describe una sal picrato con una mp de 225 °C a partir de EtOH.
DOSIFICACIÓN: 100 - 250 mg.
DURACIÓN: 6 - 8 h.