
Chicos, tengo una pregunta:
¿Hay alguna forma de obtener 3,4-metilendioxifenilpropan-2-ona (MDP2P) a partir de Eugenol?
En caso afirmativo, ¿podría alguien darme una síntesis precisa?
Encontre esta sintesis pero no entiendo como puedo hacer beta-nitroisoeugenol y β-Nitroisosafrole.
El problema es que en mi país es imposible conseguir aceite de sasafrás, safrol, isosafrol y piperonal.
En un matraz de 5 litros y 3 cuellos equipado con agitador y refrigerante de reflujo se colocaron 1000 mL de etanol y 110 g de beta-nitroisoeugenol. La mezcla se calentó con agitación y, cuando el nitroisoeugenol se disolvió, se añadieron 2500 mL de agua caliente. Con calentamiento y agitación vigorosa, se añadieron 200 g de polvo de hierro reducido y 8 g de cloruro férrico hidratado. Con agitación continuada se añadieron lentamente 100 mL de ácido clorhídrico concentrado. El ácido clorhídrico provocó una reacción violenta que remitió al cabo de unos 5 minutos; la mezcla se sometió a reflujo con agitación durante dos horas y después se destiló a presión reducida hasta recoger aproximadamente 2 litros de destilado. El residuo se filtró y el óxido de hierro esponjoso se lavó a fondo con agua caliente y después con éter. El filtrado y los lavados combinados se acidificaron fuertemente con ácido clorhídrico y se extrajeron con éter. El éter se secó y se destiló para dar un aceite ligero que se fraccionó in vacuo para dar 68 g de vanilil metil cetona que hervía a 126-127°C a 0,3 mm (rendimiento del 72%).
La 3,4-dimetoxifenilacetona y la 3,4-metilendioxifenilacetona se prepararon a partir de los β-nitropropenilbencenos correspondientes mediante procedimientos casi idénticos con un rendimiento del 90 y 72%.
Para la preparación de 2-MeO-P2P con tolueno/agua como disolvente véase Organic Syntheses, Coll. vol. 4, p. 573.
Mi modificación de esta preparación:
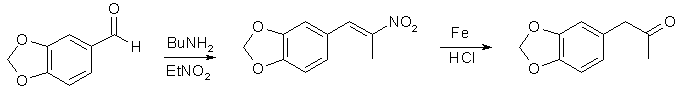
En un matraz de 2 litros y 3 cuellos equipado con agitador y refrigerante de reflujo se colocaron 460 ml de etanol1 y 67 g de β-nitroisosafrol2. La mezcla se calentó con agitación y, cuando se disolvieron los cristales amarillos, se añadieron 1100 ml de agua caliente. Con calentamiento y agitación enérgica, se añadieron 80 g de polvo de hierro reducido3 y 5 g de FeCl3-6H2O. Con agitación continuada se añadieron 63 ml de HCl conc. en 30 min. La mezcla se sometió a reflujo con agitación durante 2 h y después se destiló a presión atmosférica4. El residuo se filtró y el óxido de hierro marrón se extrajo 3x50 ml de CH2Cl2. El filtrado y los lavados orgánicos se acidificaron con HCl y se recogió la capa de cetona roja. La capa acuosa verde claro se extrajo 2x100 ml de CH2Cl2. Los extractos combinados se secaron con Na2SO4 y se destilaron a presión atmosférica (baño de 80°C). El diclorometano se eliminó completamente al vacío para dar 55 g de MDP2P crudo como aceite rojo intenso que se utilizó para la reacción CH3NH2/Al.
Notas:
¿Hay alguna forma de obtener 3,4-metilendioxifenilpropan-2-ona (MDP2P) a partir de Eugenol?
En caso afirmativo, ¿podría alguien darme una síntesis precisa?
Encontre esta sintesis pero no entiendo como puedo hacer beta-nitroisoeugenol y β-Nitroisosafrole.
El problema es que en mi país es imposible conseguir aceite de sasafrás, safrol, isosafrol y piperonal.
En un matraz de 5 litros y 3 cuellos equipado con agitador y refrigerante de reflujo se colocaron 1000 mL de etanol y 110 g de beta-nitroisoeugenol. La mezcla se calentó con agitación y, cuando el nitroisoeugenol se disolvió, se añadieron 2500 mL de agua caliente. Con calentamiento y agitación vigorosa, se añadieron 200 g de polvo de hierro reducido y 8 g de cloruro férrico hidratado. Con agitación continuada se añadieron lentamente 100 mL de ácido clorhídrico concentrado. El ácido clorhídrico provocó una reacción violenta que remitió al cabo de unos 5 minutos; la mezcla se sometió a reflujo con agitación durante dos horas y después se destiló a presión reducida hasta recoger aproximadamente 2 litros de destilado. El residuo se filtró y el óxido de hierro esponjoso se lavó a fondo con agua caliente y después con éter. El filtrado y los lavados combinados se acidificaron fuertemente con ácido clorhídrico y se extrajeron con éter. El éter se secó y se destiló para dar un aceite ligero que se fraccionó in vacuo para dar 68 g de vanilil metil cetona que hervía a 126-127°C a 0,3 mm (rendimiento del 72%).
La 3,4-dimetoxifenilacetona y la 3,4-metilendioxifenilacetona se prepararon a partir de los β-nitropropenilbencenos correspondientes mediante procedimientos casi idénticos con un rendimiento del 90 y 72%.
Para la preparación de 2-MeO-P2P con tolueno/agua como disolvente véase Organic Syntheses, Coll. vol. 4, p. 573.
Mi modificación de esta preparación:
En un matraz de 2 litros y 3 cuellos equipado con agitador y refrigerante de reflujo se colocaron 460 ml de etanol1 y 67 g de β-nitroisosafrol2. La mezcla se calentó con agitación y, cuando se disolvieron los cristales amarillos, se añadieron 1100 ml de agua caliente. Con calentamiento y agitación enérgica, se añadieron 80 g de polvo de hierro reducido3 y 5 g de FeCl3-6H2O. Con agitación continuada se añadieron 63 ml de HCl conc. en 30 min. La mezcla se sometió a reflujo con agitación durante 2 h y después se destiló a presión atmosférica4. El residuo se filtró y el óxido de hierro marrón se extrajo 3x50 ml de CH2Cl2. El filtrado y los lavados orgánicos se acidificaron con HCl y se recogió la capa de cetona roja. La capa acuosa verde claro se extrajo 2x100 ml de CH2Cl2. Los extractos combinados se secaron con Na2SO4 y se destilaron a presión atmosférica (baño de 80°C). El diclorometano se eliminó completamente al vacío para dar 55 g de MDP2P crudo como aceite rojo intenso que se utilizó para la reacción CH3NH2/Al.
Notas:
- El uso de metanol en lugar de etanol condujo a una disolución incompleta de los cristales amarillos.
- El β-nitroisosafrol puede prepararse convenientemente mediante Knoevenagel: se mezclaron 150 g de piperonal con 76 ml de nitroetano (el piperonal debe disolverse completamente para dar un líquido incoloro) y se añadieron 1,5 ml de n-butilamina(o n-pentilamina). La solución amarilla resultante se mantuvo en un lugar oscuro; empezó a aparecer algo de agua al cabo de 2-3 días a medida que avanzaba la reacción. A los 6-8 días empezaron a precipitar cristales naranja claro del producto. Después de 20 días se filtró la mezcla. El filtrado marrón claro se filtró de nuevo a los 5 días para obtener la siguiente cosecha de producto. Rendimiento 85-90% de cristales de color naranja claro, que son adecuados para la preparación de la cetona.
- Se utilizó hierro reducido con hidrógeno. El uso de polvo de hierro de malla 20 en lugar de hierro reducido condujo a una reacción menos vigorosa, el etanol resultante no contenía NH3 y la cetona estaba contaminada con aproximadamente un 20% de su oxima (reducción no cuantitativa).
- Se recogió una fracción 77-85°C. Contenía algo de NH3 que puede neutralizarse con H2SO4 conc. y destilarse de nuevo mediante columna Vigreux. El etanol al 90% resultante se utilizó para la preparación de la siguiente porción de cetona.
