2C-B
4-BROMO-2,5-DIMÉTHOXYPHÉNÉTHYLAMINE
SYNTHÈSE : Une solution de 100 g de 2,5-diméthoxybenzaldéhyde dans 220 g de nitrométhane a été traitée avec 10 g d'acétate d'ammonium anhydre et chauffée sur un bain de vapeur pendant 2,5 h en agitant de temps en temps. Le mélange réactionnel rouge foncé a été débarrassé de l'excès de nitrométhane sous vide et le résidu a cristallisé spontanément. Ce nitrostyrène brut a été purifié par broyage sous IPA, filtrage et séchage à l'air, pour donner 85 g de 2,5-diméthoxy-bêta-nitrostyrène comme produit jaune-orange d'une pureté adéquate pour l'étape suivante. Une purification supplémentaire peut être obtenue par recristallisation à partir d'IPA bouillant.
Dans un ballon à fond rond de 2 litres équipé d'un agitateur magnétique et placé sous atmosphère inerte, on a ajouté 750 ml de THF anhydre, contenant 30 g de LAH. On a ensuite ajouté, dans la solution de THF, 60 g de 2,5-diméthoxy-bêta-nitrostyrène. La solution finale était d'une couleur jaune-brun sale et a été maintenue à la température du reflux pendant 24 h. Après refroidissement, l'excès d'hydrure a été détruit par l'ajout goutte à goutte d'IPA. Ensuite, 30 ml de NaOH à 15 % ont été ajoutés pour convertir les solides inorganiques en une masse filtrable. Le mélange réactionnel a été filtré et le gâteau de filtration a été lavé d'abord avec du THF puis avec du MeOH. Les liqueurs mères et les lavages combinés ont été débarrassés du solvant sous vide et le résidu a été suspendu dans 1,5 L d'eau. Le tout a été acidifié avec HCl, lavé avec 3x100 mL de CH2Cl2, rendu fortement basique avec 25% de NaOH, et réextrait avec 4x100 mL de CH2Cl2. Les extraits réunis ont été débarrassés du solvant sous vide, ce qui a donné 26 g de résidu huileux, qui a été distillé à 120-130 °C à 0,5 mm/Hg pour donner 21 g d'une huile blanche, la 2,5-diméthoxy-phénéthylamine
(2C-H), qui absorbe très rapidement le dioxyde de carbone de l'air.
A une solution bien agitée de 24,8 g de 2,5-diméthoxyphénéthylamine dans 40 mL d'acide acétique glacial, on a ajouté 22 g de brome élémentaire dissous dans 40 mL d'acide acétique. Après quelques minutes, il y a eu formation de solides et simultanément un dégagement considérable de chaleur. Le mélange réactionnel a été ramené à la température ambiante, filtré et les solides ont été lavés avec parcimonie avec de l'acide acétique froid. Il s'agit du sel d'hydrobromure. Il existe de nombreuses formes de sel compliquées, à la fois des polymorphes et des hydrates, qui peuvent rendre l'isolement et la caractérisation du 2C-B difficiles. La voie la plus heureuse consiste à former le sel de chlorhydrate insoluble en passant par la base libre. La masse totale de sel mouillé à l'acide acétique a été dissoute dans de l'eau chaude, rendue basique à un pH d'au moins 11 avec 25% de NaOH, et extraite avec 3x100 mL de CH2Cl2. L'élimination du solvant a donné 33,7 g de résidu qui a été distillé à 115-130 °C à 0,4 mm/Hg. L'huile blanche, 27,6 g, a été dissoute dans 50 mL de H2O contenant 7,0 g d'acide acétique. Cette solution claire a été vigoureusement agitée et traitée avec 20 ml de HCl concentré. Le sel anhydre du chlorhydrate de 2,5-diméthoxy-4-bromophénéthylamine (2C-B) s'est immédiatement formé. Cette masse de cristaux a été éliminée par filtration (elle peut être considérablement détachée par l'ajout de 60 ml supplémentaires de H2O), lavée avec un peu de H2O, puis avec plusieurs portions de 50 ml d'Et2O. Après séchage complet à l'air, on obtient 31,05 g de fines aiguilles blanches, avec un mp de 237-239 °C avec décomposition. Lorsqu'il y a trop de H2O présent au moment de l'ajout du HCl concentré final, on obtient une forme hydratée du 2C-B. Le sel de bromure d'hydrogène fond. Le sel d'hydrobromure fond à 214,5-215 °C. Le sel d'acétate a été signalé comme ayant un mp de 208-209 °C.
POSOLOGIE : 12 - 24 mg.
DUREE : 4 - 8 h.
DOM
STP ; 2,5-DIMÉTHOXY-4-MÉTHYLAMPHÉTAMINE
SYNTHESE : A une solution de 54,9 g de 2,5-diméthoxy-4-méthylbenzaldéhyde (voir la recette du
2C-D pour sa préparation) dans 215 g d'acide acétique glacial, on a ajouté 19,5 g d'acétate d'ammonium anhydre et 30,6 g de nitroéthane. Ce mélange a été chauffé pendant 3 heures sur le bain de vapeur, le mélange réactionnel a été refroidi dans un bain de glace humide, ce qui a permis la formation spontanée de cristaux jaunes. Autant de H2O que possible a été ajouté (juste avant un caractère huileux trouble persistant) et après quelques heures supplémentaires de repos, le 1-(2,5-diméthoxy-4-méthylphényl)-2-nitropropène cristallin a été éliminé par filtration et recristallisé à partir d'acide acétique bouillant. Le rendement, après séchage à poids constant, était de 28,3 g et la température maximale de 87-88 °C. Anal. (C12H15NO4) C, H ,N.
Une suspension de 9,5 g de LAH dans 750 mL d'Et2O anhydre bien agité a été maintenue à reflux sous une atmosphère inerte, le retour du solvant condensé passant par une éprouvette de Soxhlet contenant 9,5 g de 1-(2,5-diméthoxy-4-méthylphényl)-2-nitropropène. Une fois l'addition du nitrostyrène terminée, la suspension agitée a été maintenue à reflux pendant 4 heures supplémentaires, puis refroidie à température ambiante et laissée sous agitation pendant une nuit. L'excès d'hydrure a été détruit par l'addition de 750 mL de H2SO4 à 8 %, prudemment, jusqu'à ce que le dégagement d'hydrogène cesse, puis à une vitesse permettant aux solides formés de se disperser. Les phases ont été séparées, la phase aqueuse a été lavée une fois avec Et2O, traitée avec 225 g de tartrate de sodium et de potassium, et enfin rendue basique (pH >9) avec 5% de NaOH. Le tout a été extrait avec 3x150 mL de CH2Cl2, les extraits ont été regroupés et le solvant a été éliminé sous vide. Le résidu était 9,6 g d'une huile claire qui a spontanément formé des cristaux avec un mp de 60,5-61 °C à partir de l'hexane. Ces solides ont été dissous dans 150 ml d'Et2O anhydre, et saturés avec du gaz HCl anhydre. Après avoir reposé à température ambiante pendant 2 h, le chlorhydrate de 2,5-diméthoxy-4-méthylamphétamine cristallin (DOM) a été éliminé par filtration, lavé avec de l'Et2O et séché à l'air jusqu'à obtention d'un poids constant. On a obtenu 8,25 g de cristaux blancs scintillants dont le mp était compris entre 190,5 et 191,5 °C. Le sulfate avait un mp de 131 °C. Anal. (C12H20ClNO2) C, H ,N.
Le nitrostyrène ci-dessus peut également être converti en produit aminé final par l'intermédiaire de la phénylacétone correspondante. A une suspension bien agitée de 10,4 g de fer en poudre dans 20 ml d'acide acétique glacial, maintenue à la température du reflux, on a ajouté 4,9 g de 1-(2,5-diméthoxy-4-méthylphényl)-2-nitropropène sous forme solide. Le reflux a été poursuivi pendant 2 heures, puis le tout a été filtré à travers de la Celite humide. Après un lavage avec 300 mL de H2O suivi de 300 mL d'Et2O, le filtrat combiné et les lavages ont été séparés, et la phase aqueuse a été extraite avec 2x100 mL d'Et2O. La phase organique et les extraits ont été combinés et lavés avec 2x100 mL de K2CO3 saturé et le solvant a été éliminé sous vide pour donner une huile rougeâtre pesant 3,3 g. Celle-ci a été distillée à 111-115 °C à 0,5 mm/Hg pour donner un solide vert pâle. Après recristallisation à partir de benzène, on a obtenu 2,8 g de 1-(2,5-diméthoxy-4-méthylphényl)-2-propanone sous forme de cristaux blancs avec un mp de 57-59 °C. Cette cétone a également été décrite comme une huile jaune pâle avec un pb de 115-118 °C à 0,4 mm/Hg. Une solution de 0,7 g de 1-(2,5-diméthoxyphényl-4-méthyl)-2-propanone dans 20 mL de MeOH a été traitée avec 6,0 g d'acétate d'ammonium, 0,3 g de cyanoborohydrure de sodium et 3 g de tamis moléculaire Linde 3 A. Le mélange a été agité pendant une nuit. Le mélange a été agité pendant une nuit, les solides ont été éliminés par filtration et le filtrat a été dissous dans 100 ml de H2O. La solution a été acidifiée avec du H2SO4 dilué, et lavée avec 2x25 mL de CH2Cl2. La phase aqueuse a été rendue basique avec du NaOH aqueux, et le produit a été extrait avec 2x25 mL de CH2Cl2. Le solvant a été éliminé sous vide et le résidu distillé (à 160 °C à 0,2 mm/Hg) pour donner un produit incolore qui a été dissous dans 3 mL d'IPA, neutralisé avec de l'HCl concentré et dilué avec 50 mL d'Et2O anhydre. On a obtenu 0,18 g de chlorhydrate de 2,5-diméthoxy-4-méthylamphétamine (DOM) sous la forme d'un solide blanc avec un mp de 187-188 °C.
Les isomères optiques de DOM ont été préparés de deux manières. La base racémique a été résolue sous forme de sel d'acide ortho-nitrotartranilique par recristallisation à partir d'EtOH. L'acide (+) fournit de préférence l'isomère (+) ou "S" de DOM. De même, la 1-(2,5-diméthoxy-4-méthylphényl)-2-propanone mentionnée ci-dessus peut être aminée de manière réductrice avec l'alpha-méthyl benzylamine optiquement active avec le nickel de Raney. Cette amine est isolée et purifiée par recristallisation du sel de chlorhydrate. Lorsqu'elle est optiquement pure, le groupe benzyle est éliminé par hydrogénolyse avec du palladium sur carbone. Le mp de l'un ou l'autre des isomères optiques, sous forme de sels de chlorhydrate, était de 204-205 °C.
POSOLOGIE : 3 - 10 mg.
DUREE : 14 - 20 h.
MDA
3,4-MÉTHYLÈNEDIOXYAMPHÉTAMINE
SYNTHESE : (à partir du pipéronal) A une solution de 15,0 g de pipéronal dans 80 ml d'acide acétique glacial, on a ajouté 15 ml de nitroéthane, puis 10 g de cyclohexylamine. Le mélange a été maintenu à la température du bain de vapeur pendant 6 heures, dilué avec 10 ml de H2O, ensemencé avec un cristal de produit et refroidi pendant une nuit à 10 °C. Les cristaux jaune vif ont été éliminés par filtration et séchés à l'air pour donner 10,7 g de 1-(3,4-méthylènedioxyphényl)-2-nitropropène avec un mp de 93-94 °C. Cette température a été portée à 97-98 °C par recristallisation à partir d'acide acétique. Les efforts plus conventionnels de synthèse du nitrostyrène utilisant un excès de nitroéthane comme solvant et de l'acétate d'ammonium anhydre comme base, donnent des produits impurs avec des rendements très faibles. Le nitrostyrène a été fabriqué avec succès à partir des composants dans du MeOH froid, avec du NaOH aqueux comme base.
Une suspension de 20 g de LAH dans 250 ml de THF anhydre a été placée sous atmosphère inerte et agitée magnétiquement. On a ajouté, goutte à goutte, 18 g de 1-(3,4-méthylènedioxyphényl)-2-nitropropène en solution dans le THF et le mélange réactionnel a été maintenu à reflux pendant 36 h. Après avoir été ramené à la température ambiante, l'hydrure en excès a été détruit avec 15 mL d'IPA, suivis de 15 mL de NaOH à 15 %. 50 mL supplémentaires de H2O ont été ajoutés pour achever la conversion des sels d'aluminium en un solide blanc, lâche, facile à filtrer. Celui-ci a été éliminé par filtration et le gâteau de filtre a été lavé avec du THF supplémentaire. Le filtrat et les lavages combinés ont été débarrassés du solvant sous vide et le résidu a été dissous dans du H2SO4 dilué. Le lavage avec 3x75 mL de CH2Cl2 a éliminé une grande partie de la couleur, et la phase aqueuse a été rendue basique et réextraite avec 3x100 mL de CH2Cl2. L'élimination du solvant a permis d'obtenir 13,0 g d'une huile de couleur jaune qui a été distillée. La fraction bouillant à 80-90 °C à 0,2 mm pesait 10,2 g et était blanche comme l'eau. Elle a été dissoute dans 60 ml d'IPA, neutralisée avec du HCl concentré et diluée avec 120 ml d'Et2O anhydre, ce qui a produit une turbidité durable. Des cristaux se sont formés spontanément et ont été éliminés par filtration, lavés avec de l'Et2O et séchés à l'air pour donner 10,4 g de chlorhydrate de 3,4-méthylènedioxyamphétamine (MDA) avec un mp de 187-188 °C.
(à partir de 3,4-méthylènedioxyphénylacétone) À une solution de 32,5 g d'acétate d'ammonium anhydre dans 120 mL de MeOH, on a ajouté 7,12 g de 3,4-méthylènedioxyphénylacétone (voir MDMA pour sa préparation), puis 2,0 g de cyanoborohydrure de sodium. La solution jaune obtenue a été vigoureusement agitée et du HCl concentré a été ajouté périodiquement pour maintenir le pH du mélange réactionnel entre 6 et 7, comme déterminé par un papier pH universel humide externe. Après plusieurs jours, des solides non dissous sont restés dans le mélange réactionnel et il n'a plus été nécessaire d'ajouter de l'acide. Le mélange réactionnel a été ajouté à 600 ml de HCl dilué, puis lavé avec 3 x 100 ml de CH2Cl2. Les lavages combinés ont été réextraits avec une petite quantité de HCl dilué, les phases aqueuses ont été combinées et rendues basiques avec 25% de NaOH. Le tout a ensuite été extrait avec 3x100 mL de CH2Cl2, ces extraits ont été combinés et le solvant a été éliminé sous vide pour fournir 3,8 g d'un résidu de couleur rouge. Celui-ci a été distillé à 80-90 °C à 0,2 mm/Hg pour fournir 2,2 g d'une huile absolument blanche comme l'eau. Il n'y a pas eu de formation évidente d'un sel de carbonate à l'exposition à l'air. Cette huile a été dissoute dans 15 mL d'IPA, neutralisée avec 25 gouttes de HCl concentré et diluée avec 30 mL d'Et2O anhydre. Lentement, des cristaux blancs de chlorhydrate de 3,4-méthylènedioxyamphétamine (MDA) se sont déposés, pesant 2,2 g et ayant un mp de 187-188 °C. La préparation du formamide (précurseur de la MDMA) et de l'acétamide (précurseur de la MDE) est décrite sous ces entrées.
POSOLOGIE : 80 - 160 mg.
DURÉE : 4 - 6
(révisé, septembre 2001).
MESCALINE ;
3,4,5-TRIMÉTHOXYPHÉNÉTHYLAMINE
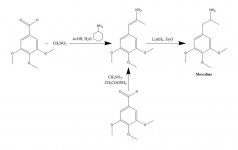
SYNTHESE : Une solution de 20 g de 3,4,5-triméthoxybenzaldéhyde, 40 mL de nitrométhane et 20 mL de cyclohexylamine dans 200 mL d'acide acétique a été chauffée au bain-marie pendant 1 h. Le mélange réactionnel a ensuite été dilué lentement et sous bonne agitation avec 400 mL de H2O, ce qui a permis la formation d'une masse cristalline jaune lourde. Celle-ci a été éliminée par filtration, lavée avec H2O et aspirée aussi sèchement que possible. La recristallisation à partir de MeOH bouillant (15 mL/g) a donné, après filtration et séchage à l'air, du bêta-nitro-3,4,5-triméthoxystyrène sous forme de cristaux jaune vif pesant 18,5 g. Une synthèse alternative a été efficace, en utilisant un excès de nitrométhane comme solvant et comme réactif, si la quantité de catalyse à l'acétate d'ammonium était maintenue à un faible niveau. Une solution de 20 g de 3,4,5-triméthoxybenzaldéhyde dans 40 mL de nitrométhane contenant 1 g d'acétate d'ammonium anhydre a été chauffée au bain-marie pendant 4 h. Le solvant a été éliminé sous vide et l'huile jaune résiduelle a été dissoute dans deux volumes de MeOH chaud, décantée de quelques insolubles et laissée à refroidir. Les cristaux formés sont éliminés par filtration, lavés avec du MeOH et séchés à l'air, ce qui donne 14,2 g de cristaux jaune vif de bêta-nitro-3,4,5-triméthoxystyrène. L'utilisation de ces proportions, mais avec 3,5 g d'acétate d'ammonium, a donné de nombreux produits de réaction secondaire, même lorsqu'ils ont été préparés après seulement 1,5 h de chauffage. Dans ce dernier cas, le rendement en nitrostyrène n'était pas satisfaisant.
A une suspension à reflux doux de 2 g de LAH dans 200 mL d'Et2O, on a ajouté 2,4 g de bêta-nitro-3,4,5-triméthoxystyrène sous forme de solution saturée d'Et2O à l'aide d'un condenseur d'extraction Soxhlet modifié pour permettre le retour continu du solvant condensé à travers le dé à coudre. Une fois l'addition terminée, les conditions de reflux ont été maintenues pendant 48 heures supplémentaires. Après refroidissement du mélange réactionnel, un total de 150 ml de H2SO4 1,5 N a été ajouté avec précaution, détruisant l'excès d'hydrure et produisant finalement deux phases claires. Celles-ci ont été séparées et la phase aqueuse a été lavée une fois avec 50 mL d'Et2O. On a ensuite ajouté 50 g de tartrate de sodium et de potassium, puis une quantité suffisante de NaOH pour amener le pH à >9. Le tout a ensuite été extrait avec 3x75 mL de CH2Cl2, et le solvant des extraits regroupés a été éliminé sous vide. Le résidu a été distillé à 120-130 °C à 0,3 mm/Hg, donnant une huile blanche qui a été dissoute dans 10 mL d'IPA et neutralisée avec de l'HCl concentré. Les cristaux blancs formés ont été dilués avec 25 mL d'Et2O, éliminés par filtration et séchés à l'air pour donner 2,1 g de chlorhydrate de 3,4,5-triméthoxyphénéthylamine (M) sous forme de cristaux blancs scintillants. Le sel de sulfate a formé des cristaux spectaculaires à partir de l'eau, mais a eu un mp large et non caractéristique. Une synthèse alternative peut utiliser le 3,4,5-triméthoxyphénylacétonitrile, comme décrit dans la rubrique bêta-D.
POSOLOGIE : 200-400 mg (sous forme de sel de sulfate), 178-356 mg (sous forme de sel de chlorhydrate)
[Note Erowid : Le texte original indiquait "178-256" mais il s'agissait d'une erreur. L'erreur a été trouvée par Bo et vérifiée avec Shulgin. Voir la page Erowid Mescaline Dosage pour
une discussion plus complète sur les formes de mescaline].
DURÉE : 10-12 h
TMA
3,4,5-TRIMÉTHOXYAMPHÉTAMINE
SYNTHESE : A une solution de 39,2 g de 3,4,5-triméthoxybenzaldéhyde dans 30 mL d'EtOH chaud, on a ajouté 15,7 g de nitroéthane, puis 1,5 mL de n-butylamine. Le mélange réactionnel a été laissé à 40 °C pendant 7 jours. Après refroidissement et grattage, on a obtenu de fines aiguilles jaunes qui, après élimination par filtration et séchage à l'air, pesaient 48 g. La recristallisation à partir d'EtOH a donné du 2-nitro-1-(3,4,5-triméthoxyphényl)propène sous forme de cristaux jaunes avec un mp de 94-95 °C. Anal. (C12H15NO5) C,H,N. Une solution de 20 g d'aldéhyde dans 75 ml de nitroéthane a été traitée avec 4 g d'acétate d'ammonium anhydre et chauffée au bain-marie jusqu'à l'obtention d'une couleur rouge foncé. L'élimination de l'excès de solvant/réactif sous vide a donné une huile rouge qui a été dissoute dans un volume égal de MeOH bouillant. En refroidissant, des cristaux jaunes de nitropropène se sont séparés. La recristallisation à partir de MeOH a donné, après séchage à l'air jusqu'à poids constant, 13,0 g avec le même mp.
Sous atmosphère inerte, 38 g de LAH ont été mouillés avec 100 mL d'Et2O anhydre, puis suspendus dans 1 L de THF sec. Le tout a été porté à un léger reflux et une solution de 43,7 g de 2-nitro-1-(3,4,5-triméthoxyphényl)propène dans 160 mL de THF a été ajoutée lentement. Le reflux a été poursuivi pendant 36 heures, puis le mélange réactionnel a été refroidi à l'aide d'un bain de glace externe. L'excès d'hydrure a été détruit par l'addition prudente de 38 mL de H2O, suivie de 38 mL de NaOH 15%, et enfin de 114 mL de H2O. Les sels inorganiques, qui auraient dû se présenter sous la forme d'une masse lâche, granuleuse et facilement filtrable, ressemblaient plutôt à une pâte de bibliothèque, mais ils ont tout de même été filtrés. Un lavage au THF a été tenté, mais il n'a pas été efficace. Le filtrat et les lavages combinés ont été débarrassés du solvant sous vide, ce qui a donné 31,5 g de la base brute sous forme d'huile ambrée. Celle-ci a été dissoute dans 140 mL d'IPA, neutralisée avec du HCl concentré (15 mL étaient nécessaires), et diluée avec 650 mL d'Et2O anhydre. On a obtenu une phase huileuse initiale qui, sous agitation continue, s'est transformée en solides rose pâle. Ceux-ci ont été finement broyés sous CH3CN pour donner 15,2 g de chlorhydrate de 3,4,5-triméthoxyamphétamine (TMA) sous forme de cristaux blancs qui ont fondu à 195-211 °C. Tous les sels d'aluminium de partout ont été dissous dans du HCl dilué, et 1 kg de tartrate de sodium et de potassium a été ajouté. L'ajout de 25% de NaOH a permis de porter le pH à >9 sans précipitation d'alumine basique. L'extraction de cette phase avec CH2Cl2, suivie de l'élimination du solvant et de la formation de sels comme décrit ci-dessus, a permis d'isoler 6,4 g supplémentaires de TMA. Le produit préparé de cette manière contient environ 10-15% de 3,5-diméthoxy-4-hydroxyamphétamine comme impureté. Une solution de 20 g de TMA obtenue de cette manière dans 200 ml de NaOH à 5 % a été extraite avec 2 x 200 ml de CH2Cl2. Les extraits regroupés ont été lavés avec 4x100 mL de NaOH à 5%, et les lavages aqueux ont été regroupés avec la phase de base originale. La phase organique a été débarrassée de son CH2Cl2 sous vide pour donner une huile qui a été dissoute dans 40 mL d'IPA, neutralisée avec du HCl concentré et diluée avec 400 mL d'Et2O anhydre. Il y a eu formation immédiate de cristaux blancs spectaculaires de chlorhydrate de 3,4,5-triméthoxyamphétamine pur, pesant 15,4 g et ayant un mp de 220-221 °C. La phase aqueuse a été ramenée à la neutralité, traitée avec 10 g de di-hydrogénophosphate de potassium, portée à un pH de 9,0 par l'addition prudente de NaOH et extraite avec 5x100 ml de CH2Cl2. L'évaporation du solvant sous vide a donné une huile qui a spontanément cristallisé. Ce produit, la 3,5-diméthoxy-4-hydroxyamphétamine, a pu être purifié par
sublimation à 130 °C à 0,2 mm/Hg. Il s'agit d'un solide cristallin blanc qui se décolore lentement à l'air. La littérature décrit un sel picrate dont le mp est de 225 °C à partir d'EtOH.
POSOLOGIE : 100 - 250 mg.
DUREE: 6 - 8 h.