2C-B
4-BROMO-2,5-DIMETOKSYFENETYLOAMINA
SYNTEZA: Roztwór 100 g 2,5-dimetoksybenzaldehydu w 220 g nitrometanu potraktowano 10 g bezwodnego octanu amonu i ogrzewano na łaźni parowej przez 2,5 h, od czasu do czasu mieszając. Głęboko czerwona mieszanina reakcyjna została usunięta z nadmiaru nitrometanu pod próżnią, a pozostałość krystalizowała spontanicznie. Surowy nitrostyren oczyszczono przez mielenie pod IPA, filtrowanie i suszenie powietrzem, uzyskując 85 g 2,5-dimetoksy-beta-nitrostyrenu jako żółto-pomarańczowy produkt o odpowiedniej czystości do następnego etapu. Dalsze oczyszczanie można osiągnąć przez rekrystalizację z wrzącego IPA.
Do kolby okrągłodennej o pojemności 2 l, wyposażonej w mieszadło magnetyczne i umieszczonej w atmosferze obojętnej, dodano 750 ml bezwodnego THF, zawierającego 30 g LAH. Następnie w roztworze THF dodano 60 g 2,5-dimetoksy-beta-nitrostyrenu. Końcowy roztwór miał brudny żółto-brązowy kolor i był utrzymywany w temperaturze wrzenia przez 24 h. Po schłodzeniu nadmiar wodorku został zniszczony przez kroplowe dodanie IPA. Następnie dodano 30 ml 15% NaOH w celu przekształcenia nieorganicznych ciał stałych w masę nadającą się do filtrowania. Mieszaninę reakcyjną przefiltrowano, a placek filtracyjny przemyto najpierw THF, a następnie MeOH. Połączone ciecze macierzyste i popłuczyny usunięto z rozpuszczalnika pod próżnią, a pozostałość zawieszono w 1,5 l H2O. Następnie zakwaszono HCl, przemyto 3x100 mL CH2Cl2, silnie zasadowym 25% NaOH i ponownie ekstrahowano 4x100 mL CH2Cl2. Połączone ekstrakty pozbawiono rozpuszczalnika pod próżnią, uzyskując 26 g oleistej pozostałości, którą oddestylowano w temperaturze 120-130°C pod ciśnieniem 0,5 mm/Hg, uzyskując 21 g białego oleju, 2,5-dimetoksy-fenyloetyloaminy
(2C-H), która bardzo szybko odbiera dwutlenek węgla z powietrza.
Do dobrze wymieszanego roztworu 24,8 g 2,5-dimetoksyfenetyloaminy w 40 ml lodowatego kwasu octowego dodano 22 g pierwiastkowego bromu rozpuszczonego w 40 ml kwasu octowego. Po kilku minutach nastąpiło tworzenie się ciał stałych i jednoczesne wydzielanie znacznej ilości ciepła. Mieszaninę reakcyjną pozostawiono do powrotu do temperatury pokojowej, przefiltrowano, a ciała stałe przemyto oszczędnie zimnym kwasem octowym. Była to sól bromowodorkowa. Istnieje wiele skomplikowanych form soli, zarówno polimorfów, jak i hydratów, które mogą sprawić, że izolacja i charakterystyka 2C-B będzie zdradliwa. Najprostszą drogą jest utworzenie nierozpuszczalnej soli chlorowodorkowej za pomocą wolnej zasady. Całą masę soli zwilżonej kwasem octowym rozpuszczono w ciepłej H2O, doprowadzono do co najmniej pH 11 za pomocą 25% NaOH i ekstrahowano 3x100 ml CH2Cl2. Po usunięciu rozpuszczalnika otrzymano 33,7 g pozostałości, którą oddestylowano w temperaturze 115-130°C pod ciśnieniem 0,4 mm/Hg. Biały olej, 27,6 g, rozpuszczono w 50 ml H2O zawierającego 7,0 g kwasu octowego. Ten klarowny roztwór energicznie mieszano i potraktowano 20 ml stężonego HCl. Natychmiast powstała bezwodna sól chlorowodorku 2,5-dimetoksy-4-bromofenyloetyloaminy (2C-B). Tę masę kryształów usunięto przez filtrację (można ją znacznie rozluźnić przez dodanie kolejnych 60 ml H2O), przemyto niewielką ilością H2O, a następnie kilkoma porcjami Et2O o objętości 50 ml. Po całkowitym wysuszeniu na powietrzu otrzymano 31,05 g drobnych białych igieł o mp 237-239 ° C z rozkładem. Gdy w czasie dodawania końcowego stężonego HCl jest zbyt dużo H2O, otrzymuje się uwodnioną formę 2C-B. Sól bromowodorkowa topi się w temperaturze 214,5-215 °C. Doniesiono, że sól octanowa ma mp 208-209 ° C.
DAWKOWANIE: 12 - 24 mg.
CZAS DZIAŁANIA: 4 - 8 h.
DOM
STP; 2,5-DIMETOKSY-4-METYLOAMFETAMINA
SYNTEZA: Do roztworu 54,9 g 2,5-dimetoksy-4-metylobenzaldehydu (patrz przepis na
2C-D ) w 215 g lodowatego kwasu octowego dodano 19,5 g bezwodnego octanu amonu i 30,6 g nitroetanu. Mieszaninę tę ogrzewano przez 3 h na łaźni parowej, mieszaninę reakcyjną ochłodzono w mokrej łaźni lodowej, umożliwiając spontaniczne tworzenie się żółtych kryształów. Dodano tyle H2O, ile było możliwe (tuż przed trwałym mętnym oleistym charakterem) i po kilku dodatkowych godzinach stania, krystaliczny 1-(2,5-dimetoksy-4-metylofenylo)-2-nitropropen usunięto przez filtrację i rekrystalizowano z wrzącego kwasu octowego. Wydajność, po wysuszeniu do stałej masy, wynosiła 28,3 g, a mp 87-88 °C. Anal. (C12H15NO4) C, H, N.
Zawiesinę 9,5 g LAH w 750 ml dobrze mieszanego bezwodnego Et2O utrzymywano pod chłodnicą zwrotną w atmosferze obojętnej, przy czym powrót skroplonego rozpuszczalnika przechodził przez gilzę Soxhleta zawierającą 9,5 g 1-(2,5-dimetoksy-4-metylofenylo)-2-nitropropenu. Po zakończeniu dodawania nitrostyrenu, mieszaną zawiesinę utrzymywano w temperaturze wrzenia przez dodatkowe 4 godziny, a następnie schłodzono do temperatury pokojowej i pozostawiono do dalszego mieszania przez noc. Nadmiar wodorku został zniszczony przez dodanie 750 ml 8% H2SO4, ostrożnie, aż do ustania wydzielania wodoru, a następnie z prędkością, która pozwoliła na zdyspergowanie utworzonych ciał stałych. Fazy rozdzielono, fazę wodną przemyto raz Et2O, potraktowano 225 g winianu sodowo-potasowego, a na koniec zasadowano (pH >9) 5% NaOH. Następnie ekstrahowano 3x150 ml CH2Cl2, ekstrakty połączono, a rozpuszczalnik usunięto pod próżnią. Pozostałością było 9,6 g klarownego oleju, który spontanicznie utworzył kryształy o mp 60,5-61 ° C z heksanu. Te ciała stałe rozpuszczono w 150 ml bezwodnego Et2O i nasycono bezwodnym gazowym HCl. Po odstawieniu w temperaturze pokojowej na 2 godziny, krystaliczny chlorowodorek 2,5-dimetoksy-4-metyloamfetaminy (DOM) usunięto przez filtrację, przemyto Et2O i wysuszono na powietrzu do stałej masy. Otrzymano 8,25 g lśniących białych kryształów o mp 190,5-191,5 °C. Siarczan miał mp 131 °C. Analiza. (C12H20ClNO2) C, H, N.
Powyższy nitrostyren można również przekształcić w końcowy produkt aminowy poprzez pośrednictwo odpowiedniego fenyloacetonu. Do dobrze wymieszanej zawiesiny 10,4 g sproszkowanego żelaza w 20 ml lodowatego kwasu octowego utrzymywanej w temperaturze wrzenia dodano 4,9 g 1-(2,5-dimetoksy-4-metylofenylo)-2-nitropropenu w postaci ciała stałego. Refluksowanie kontynuowano przez 2 godziny, a następnie całość przefiltrowano przez mokry Celite. Po przemyciu 300 ml H2O, a następnie 300 ml Et2O, połączony filtrat i popłuczyny oddzielono, a fazę wodną ekstrahowano 2x100 ml Et2O. Fazę organiczną i ekstrakty połączono i przemyto 2x100 mL nasyconego K2CO3, a rozpuszczalnik usunięto pod próżnią uzyskując czerwonawy olej o masie 3,3 g. Destylowano go w temperaturze 111-115 °C pod ciśnieniem 0,5 mm/Hg uzyskując bladozielone ciało stałe. Po rekrystalizacji z benzenu otrzymano 2.8 g 1-(2,5-dimetoksy-4-metylofenylo)-2-propanonu w postaci białych kryształów o mp 57-59 °C. Keton ten został również opisany jako bladożółty olej o bp 115-118 °C przy 0,4 mm/Hg. Roztwór 0,7 g 1-(2,5-dimetoksyfenylo-4-metylo)-2-propanonu w 20 ml MeOH potraktowano 6,0 g octanu amonu, 0,3 g cyjanoborowodorku sodu i 3 g sita molekularnego Linde 3 A. Mieszaninę mieszano przez noc. Mieszaninę mieszano przez noc, ciała stałe usunięto przez filtrację, a filtrat rozpuszczono w 100 ml H2O. Roztwór zakwaszono rozcieńczonym H2SO4 i przemyto 2x25 mL CH2Cl2. Fazę wodną zobojętniono wodnym NaOH, a produkt ekstrahowano 2x25 mL CH2Cl2. Rozpuszczalnik usunięto pod próżnią, a pozostałość oddestylowano (w 160°C przy 0,2 mm/Hg) otrzymując bezbarwny produkt, który rozpuszczono w 3 ml IPA, zneutralizowano stężonym HCl i rozcieńczono 50 ml bezwodnego Et2O. Otrzymano 0,18 g chlorowodorku 2,5-dimetoksy-4-metyloamfetaminy (DOM) jako białe ciało stałe o mp 187-188 °C.
Izomery optyczne DOM zostały przygotowane na dwa sposoby. Zasadę racemiczną rozdzielono jako sól kwasu orto-nitrotartranilowego przez rekrystalizację z EtOH. Kwas (+) zapewnia preferencyjnie izomer (+) lub "S" DOM. Ponadto, wyżej wspomniany 1-(2,5-dimetoksy-4-metylofenylo)-2-propanon może być redukcyjnie aminowany optycznie czynną alfa-metylobenzyloaminą za pomocą niklu Raneya. Amina ta jest izolowana i oczyszczana przez rekrystalizację soli chlorowodorkowej. Po uzyskaniu czystości optycznej grupę benzylową usunięto przez uwodornienie za pomocą palladu na węglu. Mp każdego z izomerów optycznych, jako soli chlorowodorku, wynosiła 204-205 ° C.
DAWKOWANIE: 3 - 10 mg.
CZAS TR WANIA: 14 - 20 h.
MDA
3,4-METYLENODIOKSYAMFETAMINA
SYNTEZA: (z piperonalu) Do roztworu 15,0 g piperonalu w 80 ml lodowatego kwasu octowego dodano 15 ml nitroetanu, a następnie 10 g cykloheksyloaminy. Mieszaninę utrzymywano w temperaturze łaźni parowej przez 6 godzin, rozcieńczono 10 ml H2O, posiano kryształ produktu i schłodzono przez noc w temperaturze 10°C. Jasnożółte kryształy usunięto przez filtrację i wysuszono na powietrzu, uzyskując 10,7 g 1-(3,4-metylenodioksyfenylo)-2-nitropropenu o mp 93-94 °C. Temperatura ta wzrosła do 97-98 °C. Temperaturę tę zwiększono do 97-98°C przez rekrystalizację z kwasu octowego. Bardziej konwencjonalne próby syntezy nitrostyrenu przy użyciu nadmiaru nitroetanu jako rozpuszczalnika i bezwodnego octanu amonu jako zasady dają nieczysty produkt z bardzo niską wydajnością. Nitrostyren został z powodzeniem wytworzony ze składników w zimnym MeOH, z wodnym NaOH jako zasadą.
Zawiesinę 20 g LAH w 250 ml bezwodnego THF umieszczono w atmosferze obojętnej i mieszano magnetycznie. Dodano kroplami 18 g 1-(3,4-metylenodioksyfenylo)-2-nitropropenu w roztworze w THF i utrzymywano mieszaninę reakcyjną pod chłodnicą zwrotną przez 36 h. Po przywróceniu do temperatury pokojowej nadmiar wodorku zniszczono 15 ml IPA, a następnie 15 ml 15% NaOH. Dodano dodatkowe 50 ml H2O, aby zakończyć konwersję soli glinu do luźnego, białego, łatwego do przefiltrowania ciała stałego. Zostało ono usunięte przez filtrację, a placek filtracyjny przemyto dodatkowym THF. Połączony filtrat i popłuczyny usunięto z rozpuszczalnika pod próżnią, a pozostałość rozpuszczono w rozcieńczonym H2SO4. Przemywanie 3x75 mL CH2Cl2 usunęło znaczną część koloru, a faza wodna stała się zasadowa i ponownie ekstrahowana 3x100 mL CH2Cl2. Usunięcie rozpuszczalnika dało 13,0 g żółtego oleju, który poddano destylacji. Frakcja wrząca w temperaturze 80-90 °C przy 0,2 mm ważyła 10,2 g i była biało-wodna. Rozpuszczono ją w 60 ml IPA, zneutralizowano stężonym HCl i rozcieńczono 120 ml bezwodnego Et2O, co spowodowało trwałe zmętnienie. Spontanicznie utworzyły się kryształy, które usunięto przez filtrację, przemyto Et2O i wysuszono na powietrzu, uzyskując 10,4 g chlorowodorku 3,4-metylenodioksyamfetaminy (MDA) o mp 187-188 °C.
(z 3,4-metylenodioksyfenyloacetonu) Do roztworu 32,5 g bezwodnego octanu amonu w 120 ml MeOH dodano 7,12 g 3,4-metylenodioksyfenyloacetonu (sposób przygotowania patrz MDMA), a następnie 2,0 g cyjanoborowodorku sodu. Powstały żółty roztwór energicznie mieszano i okresowo dodawano stężony HCl, aby utrzymać pH mieszaniny reakcyjnej między 6 a 7, jak określono za pomocą zewnętrznego wilgotnego uniwersalnego papierka pH. Po kilku dniach w mieszaninie reakcyjnej pozostały nierozpuszczone ciała stałe i nie było już potrzeby dodawania kwasu. Mieszaninę reakcyjną dodano do 600 ml rozcieńczonego HCl, a następnie przemyto 3x100 ml CH2Cl2. Połączone popłuczyny poddano ekstrakcji zwrotnej niewielką ilością rozcieńczonego HCl, połączono fazy wodne i doprowadzono do stanu zasadowego za pomocą 25% NaOH. Następnie ekstrahowano 3x100 ml CH2Cl2, ekstrakty te połączono, a rozpuszczalnik usunięto pod próżnią, uzyskując 3,8 g czerwonej pozostałości. Pozostałość oddestylowano w temperaturze 80-90°C pod ciśnieniem 0,2 mm/Hg, uzyskując 2,2 g całkowicie wodnistego oleju. Nie stwierdzono wyraźnego tworzenia się soli węglanowej po wystawieniu na działanie powietrza. Rozpuszczono go w 15 ml IPA, zneutralizowano 25 kroplami stężonego HCl i rozcieńczono 30 ml bezwodnego Et2O. Powoli osadzały się białe kryształy chlorowodorku 3,4-metylenodioksyamfetaminy (MDA), który ważył 2,2 g i miał mp 187-188 ° C. Przygotowanie formamidu (prekursora MDMA) i acetamidu (prekursora MDE) opisano w tych pozycjach.
DAWKOWANIE: 80 - 160 mg.
CZAS TR WANIA: 4 - 6
(poprawione, wrzesień 2001).
MESCALINE;
3,4,5-TRIMETOKSYFENETYLOAMINA
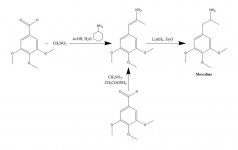
SYNTEZA: Roztwór 20 g 3,4,5-trimetoksybenzaldehydu, 40 mL nitrometanu i 20 mL cykloheksyloaminy w 200 mL kwasu octowego ogrzewano na łaźni parowej przez 1 h. Mieszaninę reakcyjną następnie powoli rozcieńczano, dobrze mieszając, 400 mL H2O, co pozwoliło na utworzenie ciężkiej żółtej krystalicznej masy. Usunięto ją przez filtrację, przemyto H2O i odessano do sucha. Rekrystalizacja z wrzącego MeOH (15 mL/g) dała, po filtracji i wysuszeniu na powietrzu, beta-nitro-3,4,5-trimetoksystyren w postaci jasnożółtych kryształów o masie 18,5 g. Alternatywna synteza była skuteczna, przy użyciu nadmiaru nitrometanu jako rozpuszczalnika, jak również odczynnika, jeśli ilość katalizatora octanu amonu była utrzymywana na niskim poziomie. Roztwór 20 g 3,4,5-trimetoksybenzaldehydu w 40 ml nitrometanu zawierającego 1 g bezwodnego octanu amonu ogrzewano na łaźni parowej przez 4 h. Rozpuszczalnik usunięto pod próżnią, a pozostały żółty olej rozpuszczono w dwóch objętościach gorącego MeOH, zdekantowano z niektórych nierozpuszczalników i pozostawiono do ostygnięcia. Powstałe kryształy usunięto przez filtrację, przemyto MeOH i wysuszono na powietrzu, uzyskując 14,2 g jasnożółtych kryształów beta-nitro-3,4,5-trimetoksystyrenu. Zastosowanie tych proporcji, ale z 3,5 g octanu amonu, dało rozległe produkty reakcji ubocznych, nawet po obróbce po zaledwie 1,5 h ogrzewania. Wydajność nitrostyrenu była w tym ostatnim przypadku niezadowalająca.
Do delikatnie refluksującej zawiesiny 2 g LAH w 200 ml Et2O dodano 2,4 g beta-nitro-3,4,5-trimetoksystyrenu w postaci nasyconego roztworu Et2O przy użyciu skraplacza ekstrakcyjnego Soxhleta zmodyfikowanego tak, aby umożliwić ciągły powrót skroplonego rozpuszczalnika przez gilzę. Po zakończeniu dodawania warunki wrzenia utrzymywano przez kolejne 48 h. Po schłodzeniu mieszaniny reakcyjnej ostrożnie dodano łącznie 150 ml 1,5 N H2SO4, niszcząc nadmiar wodorku i ostatecznie uzyskując dwie klarowne fazy. Zostały one rozdzielone, a fazę wodną przemyto raz 50 ml Et2O. Następnie dodano 50 g winianu sodowo-potasowego, a następnie wystarczającą ilość NaOH, aby uzyskać pH >9. Następnie ekstrahowano 3x75 mL CH2Cl2, a rozpuszczalnik z połączonych ekstraktów usunięto pod próżnią. Pozostałość oddestylowano w temperaturze 120-130°C pod ciśnieniem 0,3 mm/Hg, uzyskując biały olej, który rozpuszczono w 10 ml IPA i zneutralizowano stężonym HCl. Powstałe białe kryształy rozcieńczono 25 ml Et2O, usunięto przez filtrację i wysuszono na powietrzu, uzyskując 2,1 g chlorowodorku 3,4,5-trimetoksyfenyloetyloaminy (M) w postaci lśniących białych kryształów. Sól siarczanowa tworzyła spektakularne kryształy z wody, ale miała szerokie i nietypowe mp. Alternatywna synteza może wykorzystywać 3,4,5-trimetoksyfenyloacetonitryl, jak opisano w beta-D.
DAWKOWANIE: 200-400 mg (jako sól siarczanowa), 178-356 mg (jako sól chlorowodorkowa)
[Uwaga Erowid: Oryginalny tekst brzmiał "178-256", ale był to błąd. Błąd został znaleziony przez Bo i zweryfikowany przez Shulgina. Zobacz stronę Erowid Mescaline Dosage, aby uzyskać
pełniejsze omówienie form meskaliny].
CZAS TRWANIA: 10-12 h
TMA
3,4,5-TRIMETOKSYAMFETAMINA
SYNTEZA: Do roztworu 39,2 g 3,4,5-trimetoksybenzaldehydu w 30 ml ciepłego EtOH dodano 15,7 g nitroetanu, a następnie 1,5 ml n-butyloaminy. Mieszaninę reakcyjną pozostawiono w temperaturze 40°C na 7 dni. Po schłodzeniu i zdrapaniu otrzymano drobne żółte igły, które po usunięciu przez filtrację i suszenie powietrzem ważyły 48 g. Rekrystalizacja z EtOH dała 2-nitro-1-(3,4,5-trimetoksyfenylo)propen w postaci żółtych kryształów o mp 94-95 ° C. Anal. (C12H15NO5) C,H,N. Alternatywnie, roztwór 20 g aldehydu w 75 ml nitroetanu potraktowano 4 g bezwodnego octanu amonu i ogrzewano na łaźni parowej do uzyskania głębokiego czerwonego koloru. Usunięcie nadmiaru rozpuszczalnika/odczynnika pod próżnią dało czerwony olej, który rozpuszczono w takiej samej objętości wrzącego MeOH. Po schłodzeniu oddzieliły się żółte kryształy nitropropenu. Rekrystalizacja z MeOH dała, po wysuszeniu na powietrzu do stałej masy, 13,0 g o tej samej mp.
W atmosferze obojętnej 38 g LAH zwilżono 100 ml bezwodnego Et2O, a następnie zawieszono w 1 l suchego THF. Doprowadzono do łagodnego wrzenia i powoli dodano 43,7 g roztworu 2-nitro-1-(3,4,5-trimetoksyfenylo)propenu w 160 ml THF. Refluksowanie kontynuowano przez 36 h, a następnie mieszaninę reakcyjną chłodzono w zewnętrznej łaźni lodowej. Nadmiar wodorku został zniszczony przez ostrożne dodanie 38 ml H2O, a następnie 38 ml 15% NaOH, a na koniec kolejne 114 ml H2O. Sole nieorganiczne, które powinny skończyć jako luźna, ziarnista, łatwa do przefiltrowania masa, wyglądały raczej jak pasta biblioteczna, ale mimo to zostały przefiltrowane. Próbowano przemyć THF, ale nie było to skuteczne. Połączony filtrat i popłuczyny usunięto z rozpuszczalnika pod próżnią, uzyskując 31,5 g surowej zasady w postaci bursztynowego oleju. Został on rozpuszczony w 140 ml IPA, zneutralizowany stężonym HCl (wymagane było 15 ml) i rozcieńczony 650 ml bezwodnego Et2O. Pojawiła się początkowa faza oleista, która po dalszym mieszaniu zmieniła się w bladoróżowe ciała stałe. Zostały one drobno zmielone pod CH3CN, dając 15,2 g chlorowodorku 3,4,5-trimetoksyamfetaminy (TMA) w postaci białych kryształów, które topiły się w temperaturze 195-211°C. Wszystkie sole glinu rozpuszczono w rozcieńczonym HCl i dodano 1 kg winianu sodowo-potasowego. Dodanie 25% NaOH pozwoliło na doprowadzenie pH do >9 bez wytrącania zasadowego tlenku glinu. Ekstrakcja tej fazy za pomocą CH2Cl2, a następnie usunięcie rozpuszczalnika i utworzenie soli, jak opisano powyżej, pozwoliło na wyizolowanie dodatkowych 6,4 g TMA. Produkt przygotowany w ten sposób zawiera około 10-15% 3,5-dimetoksy-4-hydroksyamfetaminy jako zanieczyszczenie. Roztwór 20 g TMA przygotowany w ten sposób w 200 ml 5% NaOH ekstrahowano 2x200 ml CH2Cl2. Połączone ekstrakty przemyto 4x100 ml 5% NaOH, a wodne popłuczyny połączono z oryginalną fazą podstawową. Fazę organiczną pozbawiono CH2Cl2 pod próżnią, uzyskując olej, który rozpuszczono w 40 ml IPA, zneutralizowano stężonym HCl i rozcieńczono 400 ml bezwodnego Et2O. Natychmiast powstały spektakularne białe kryształy czystego chlorowodorku 3,4,5-trimetoksyamfetaminy o masie 15,4 g i mp 220-221°C. Fazę wodną doprowadzono do obojętności, potraktowano 10 g diwodorofosforanu potasu, doprowadzono do pH 9,0 przez ostrożne dodanie NaOH i ekstrahowano 5x100 mL CH2Cl2. Odparowanie rozpuszczalnika pod próżnią dało olej, który spontanicznie krystalizował. Produkt ten, 3,5-dimetoksy-4-hydroksyamfetamina, może być dalej oczyszczany przez
sublimację w temperaturze 130 °C przy 0,2 mm/Hg. Było to białe krystaliczne ciało stałe, które powoli odbarwiało się w powietrzu. Literatura opisuje sól pikrynianu o mp 225 ° C z EtOH.
DAWKOWANIE: 100 - 250 mg.
CZAS TR WANIA: 6 - 8 h.