
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 2,659
- Solutions
- 3
- Reaction score
- 2,734
- Points
- 113
- Deals
- 1
Introdução
O ácido lisérgico, o fragmento básico derivado dos alcaloides do ergot, foi sintetizado em uma sequência de quatorze, começando com o 3-beta-carboxietilindole. O material inicial foi convertido no intermediário 1-benzoil-5-ceto-1,2,2a-3,4,5-hexa-hidrobenz-[cd]-indole (3), que contém três dos quatro anéis presentes no ácido lisérgico. Essa cetona, por sua vez, foi transformada no composto tetracíclico, 9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fe]-quinolina (8), e daí para o ácido lisérgico (14). Essa síntese não é simples e requer muita experiência em laboratório e conhecimento de química. Além disso, há várias manipulações com substâncias perigosas, que precisam ser realizadas com medidas de segurança rigorosas.
Ponto de ebulição: 536,2±50,0 °C a 760 mm Hg;
Ponto de fusão: 240 °C;
Peso molecular: 268,31 g/mole;
Densidade: 1,4 ± 0,1 g/mL;
Número CAS: 82-58-6.
Equipamentos e vidraria:
- Reator de hidrogenação de aço de 2-3 L;
- Autoclave de aço de 500 mL;
- Balança de laboratório (0,01 - 500 g é adequado);
- Frascos de fundo redondo de 100, 200, 500 mL, 5 e 10 L;
- Compressor de hidrogênio (H2) e origem;
- Balão de Buchner e funil (grande) de 5 L [o filtro Schott pode ser usado para pequenas quantidades];
- Máquina Rotovap (grande);
- Fonte de vácuo;
- Funis de separação de 500 mL e 2 L;
- Balão de nitrogênio ~50-70 L (1 bar);
- Tampas de septo para frascos;
- Banho de água com gelo e sal;
- 5 L x2, 2 L x2; 1 L x2; 500 mL x2; 100 mL x3; 50 mL x2 Béqueres;
- Seringa de vidro ou pipeta Pasteur;
- Agitador magnético ou agitador de topo;
- Configuração de destilação a vácuo;
- Condensador de refluxo;
- Suporte de retorta e grampo para fixar o aparelho;
- Termômetro de laboratório (-20 °C a 200 °C) com adaptador de frasco;
- Papel indicador de pH;
- Bastão de vidro e espátula;
- Lâmpada de 250 watts.
Reagentes.
- Ácido 3-indolepropiônico (1), 94,6 g (0,5 mol);
- 9,5 L de água destilada (H2O);
- ~400 g de hidróxido de sódio (NaOH);
- 116 g de níquel Raney (Ni);
- 1050 mL de ácido clorídrico (HCl) concentrado;
- 2 mL de ácido sulfúrico (H2SO4 conc.);
- 210 mL de solução aq. de hidróxido de sódio (NaOH) 12N;
- 180 mL de cloreto de benzoíla;
- ~1,5 L de metanol (MeOH);
- ~1,6 L de etanol (EtOH);
- 201,2 mL de cloreto de tionila (SOCl2);
- 1950 mL de dissulfeto de carbono (CS2);
- 240 g de cloreto de alumínio (AlCl3);
- 2,5 L de benzeno;
- 500 mL de hidróxido de sódio 2N (NaOH);
- ~3,2 L de éter dietílico (Et2O);
- 3,3 L de ácido acético glacial (AcOH);
- 352 g (1,1 mol) de perbrometo de piridina;
- 5 L Clorofórmio (CHCl3);
- ~1000 g de sulfato de magnésio (MgSO4);
- 307 g (2,35 mol) Metilaminoacetona etileno-cetal (5);
- 4,5 L Benzeno;
- ~500 g de carvão ativado (C);
- ~1 L de acetona;
- ~500 g de bicarbonato de sódio (NaHCO3);
- 80 mL de anidrido acético frio (Ac2O);
- 1,5 g de borohidreto de sódio (NaBH4);
- 75 ml de dióxido de enxofre (SO2 líquido);
- 40 g de cianeto de sódio (NaCN em pó);
- 300 mL de cianeto de hidrogênio (HCN líquido);
- 78 mL de solução aq. de hidróxido de potássio (KOH) a 1,5%;
- 8,5 g de arseniato de sódio hidratado;
- ~ 50 mL de xileno;
- 100 mL de solução diluída de hidróxido de amônio (NH4OH);
- 16,9 g de metóxido de sódio (MeONa).
Procedimento
1-Benzoil-3-(beta-carboxietil)-2,3-diidroindol (2)O ácido 3-indolepropiônico (1), 94,6 g (0,5 mol), foi dissolvido em 600 mL de água contendo 20 g de hidróxido de sódio. A solução foi misturada com cerca de 100 g de catalisador de níquel Raney e hidrogenada à temperatura ambiente em uma bomba de hidrogenação de aço de 2-3 L a uma pressão de H2 de 3000-4000 psi (207-276 bar). Em geral, a redução foi concluída em 20 a 30 horas, após as quais o catalisador foi filtrado e lavado com um pouco de água. O ácido HCl concentrado, 85 ml, foi adicionado ao filtrado e a solução foi resfriada. Se a redução foi incompleta, o ácido indolepropiônico que não reagiu se separou nesse ponto e foi removido por filtração. O filtrado foi então benzoilado pelo procedimento usual de Schotten-Baumann, usando 210 mL de hidróxido de sódio 12N e 180 mL de cloreto de benzoíla. A solução foi mantida alcalina durante toda a benzoilação, e a temperatura foi mantida abaixo de 40 °C por meio de resfriamento. Quando o cloreto de benzoíla reagiu completamente, a mistura foi resfriada e acidificada com 300 mL de ácido HCl concentrado. O produto bruto foi filtrado e lavado com água, após o que foi extraído com 4 porções de 1 L de água quente para remover o ácido benzoico. O produto xaroposo quente (2), após a decantação do extrato aquoso, foi cristalizado a partir de alguns volumes de metanol; rendimento de 103 g (70 %), MP: 151-153 °C.
1-Benzoil-5-ceto-1,2,2a,3,4,5-hexahidrobenz-[cd]-indole (3)
1-Benzoil-3-(beta-carboxietil)-2,3-diidroindole (2), 118 g (0,4 mol), foi misturado com 200 mL de cloreto de tionila puro. A solução foi deixada em repouso por 30 minutos, após o que foi aquecida suavemente por 15 a 20 minutos em um banho de vapor. O excesso de cloreto de tionila foi completamente evaporado abaixo de 30 °C no vácuo, e o cloreto de ácido bruto foi dissolvido em 200 mL de dissulfeto de carbono. A solução de cloreto de ácido foi então adicionada em um fluxo fino a uma suspensão vigorosamente agitada de 240 g de cloreto de alumínio em 1.750 mL de dissulfeto de carbono contido em um balão de 5 L (no HOOD!!!). Um complexo se separou, e a agitação se tornou difícil. A mistura foi aquecida sob refluxo e agitada por uma hora para completar a reação, após o que foi decomposta com muito cuidado pela adição de 500 g de gelo, 250 mL de ácido HCl conc. e 500 mL de água. Durante a decomposição, a agitação foi mantida e o resfriamento foi afetado pela destilação periódica do dissulfeto de carbono no vácuo, e o produto foi extraído com 2 L de benzeno. O extrato foi lavado completamente com 500 mL de hidróxido de sódio 2N em três porções e depois com água. Ele foi seco sobre sulfato de magnésio e evaporado a um pequeno volume no vácuo. A adição lenta de vários volumes de éter causou a cristalização da cetona amarela (3) . Ela foi filtrada e lavada com éter; rendimento de 85,3 g (77%), MP: 146-147 °C. Uma amostra foi recristalizada para análise a partir do éter de benzeno.
1-Benzoil-3-(beta-carboxietil)-2,3-diidroindole (2), 118 g (0,4 mol), foi misturado com 200 mL de cloreto de tionila puro. A solução foi deixada em repouso por 30 minutos, após o que foi aquecida suavemente por 15 a 20 minutos em um banho de vapor. O excesso de cloreto de tionila foi completamente evaporado abaixo de 30 °C no vácuo, e o cloreto de ácido bruto foi dissolvido em 200 mL de dissulfeto de carbono. A solução de cloreto de ácido foi então adicionada em um fluxo fino a uma suspensão vigorosamente agitada de 240 g de cloreto de alumínio em 1.750 mL de dissulfeto de carbono contido em um balão de 5 L (no HOOD!!!). Um complexo se separou, e a agitação se tornou difícil. A mistura foi aquecida sob refluxo e agitada por uma hora para completar a reação, após o que foi decomposta com muito cuidado pela adição de 500 g de gelo, 250 mL de ácido HCl conc. e 500 mL de água. Durante a decomposição, a agitação foi mantida e o resfriamento foi afetado pela destilação periódica do dissulfeto de carbono no vácuo, e o produto foi extraído com 2 L de benzeno. O extrato foi lavado completamente com 500 mL de hidróxido de sódio 2N em três porções e depois com água. Ele foi seco sobre sulfato de magnésio e evaporado a um pequeno volume no vácuo. A adição lenta de vários volumes de éter causou a cristalização da cetona amarela (3) . Ela foi filtrada e lavada com éter; rendimento de 85,3 g (77%), MP: 146-147 °C. Uma amostra foi recristalizada para análise a partir do éter de benzeno.
1-Benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (4)
1-Benzoil-2,2a,3,4-tetraidro-4-[metil-(2-metil-1,2-dioxolan-2-il-metil)-amino]-benz-[cd]-indol-5-(1H)-ona (6)
Uma solução de 270 g (0,76 mol) de 1-benzoil-4-bromo-5-ceto-1,2,2a,3,4,5-hexa-hidrobenz-[cd]-indole(4) e 307 g (2.35 mol) de etileno-cetal de metilaminoacetona (5) em 4.500 mL de benzeno seco foram refluxados sob nitrogênio por 21 h em 10 L RBF com condensador de refluxo. A mistura foi resfriada, e 151 g (93,5%) de hidrobrometo de metilaminoacetona etileno-cetal foram filtrados, MP: 158-159 °C.
O filtrado foi lavado várias vezes com água gelada, após o que foi extraído com 2,5 L de ácido HCl diluído frio contendo 150 mL do ácido concentrado. Os extratos ácidos foram imediatamente adicionados a um excesso de hidróxido de sódio diluído e gelado. O produto foi extraído com 1 L de clorofórmio, e a solução de clorofórmio foi seca sobre sulfato de magnésio, tratada com carbono e concentrada no vácuo. A cetal-cetona residual (6) foi cristalizada a partir de acetona; MP: e mistura MP: 135-136 °C, com rendimento de 220 g (71%).
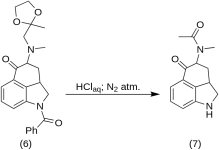
5-Keto-4-[N-metil-N-acetonilamino]-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (7)
20 g de 1-benzoil-2,2a,3,4-tetrahidro-4-[metil-(2-metil-1,3-dioxolan-2-il-metil)-amino]-benz-[cd]-indol-5-(1H)-ona (6) foi dissolvido em uma mistura de 250 mL de ácido HCl concentrado e 250 mL de água, e a solução foi mantida sob nitrogênio a 37 °C em 3-5 L RBF por cinco dias. A mistura foi resfriada, tratada com carbono, filtrada e o filtrado foi concentrado no vácuo até um pequeno volume. O resíduo foi tratado com excesso de bicarbonato de sódio; o produto foi extraído com clorofórmio frio, e o solvente foi removido no vácuo à temperatura ambiente. A dicetona bruta (7) foi pulverizada, misturada com cerca de 75 mL de éter benzênico 1:1 e filtrada; rendimento de 9,8 g (77%), MP: 105-107 °C. Uma amostra para análise foi recristalizada a partir de éter de benzeno ou etanol; MP: 109-110 °C; um monocloridrato foi obtido a partir de etanol diluído; MP: 200 °C dec.
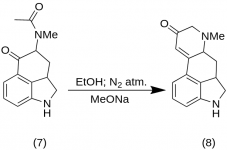
9-Keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (8)
25 g de 5-Keto-4-[N-methyl-N-acetonyl]-amino-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (7) foram misturados com 550 mL de etanol absoluto. A mistura foi agitada sob nitrogênio e resfriada a -15 °C em 2-5 L de RBF. Em seguida, foi adicionado metóxido de sódio, 16,9 g, e a mistura foi agitada entre -10 °C e -12 °C por dez minutos. A mistura de reação foi resfriada a -25 °C, e o produto foi filtrado em um funil de Buchner de 6,5 polegadas e lavado com um pouco de etanol frio e éter. Com o mínimo de exposição ao ar (contém metóxido de sódio!), a cetona bruta (8) foi imediatamente agitada com um pouco de água gelada e filtrada novamente. Ela foi lavada com água gelada, etanol e éter; rendimento de 16,2 g (69%), MP: 145-147 °C. Uma amostra analítica foi recristalizada a partir de etanol diluído; MP: 155-157 °C; O dicloridrato foi preparado e recristalizado a partir de acetona aquosa; MP: 270 °C dec.
4-Acetil-9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (9)
9-Ceto-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (8), 24 g, foi adicionado a 80 mL de anidrido acético frio. A mistura foi mantida a 25 °C em 200 mL de RBF por cerca de 5 min, após o que foi completamente resfriada, e o produto (9) foi filtrado e lavado com éter; rendimento de 20,5 g (76 %), mp: 167-170 °C. Uma segunda colheita foi obtida por evaporação do filtrado, o que aumentou o rendimento total para 82%. Uma amostra foi recristalizada a partir de acetona-etanol; MP: 169-170 °C; O cloridrato foi preparado em etanol e foi recristalizado a partir de etanol aquoso; MP: 250 °C dec.
9-Ceto-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (8), 24 g, foi adicionado a 80 mL de anidrido acético frio. A mistura foi mantida a 25 °C em 200 mL de RBF por cerca de 5 min, após o que foi completamente resfriada, e o produto (9) foi filtrado e lavado com éter; rendimento de 20,5 g (76 %), mp: 167-170 °C. Uma segunda colheita foi obtida por evaporação do filtrado, o que aumentou o rendimento total para 82%. Uma amostra foi recristalizada a partir de acetona-etanol; MP: 169-170 °C; O cloridrato foi preparado em etanol e foi recristalizado a partir de etanol aquoso; MP: 250 °C dec.
4-Acetil-9-hidroxi-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (10)
10 g de 4-acetil-9-ceto-7-metil-4,5,5a,6,6a,7,8,9-octahidroxindolo-[4,3-fg]-quinolina (9) foram adicionados a uma mistura de 150 mL de metanol e 10 mL de água em 500 mL de RBF. Adicionou-se borohidreto de sódio, 1,5 g, e deixou-se a reação prosseguir à temperatura ambiente até um pequeno volume, e adicionou-se uma mistura de 15 mL de ácido HCl conc. e 60 mL de água. O cloridrato (10) que se separou no resfriamento foi filtrado e lavado com metanol, 9,0 g (79%). Uma amostra foi recristalizada a partir de etanol diluído; MP: 245-246 °C dec.
Cloridrato de 4-acetil-9-cloro-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (11)
Cloridrato de 4-acetil-9-hidroxi-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (10), 3.1 g, foi dissolvido em 75 mL de dióxido de enxofre líquido contido em um revestimento de vidro em uma autoclave de aço de 500 mL. Adicionou-se 1,2 mL de cloreto de tionila e o recipiente foi vedado e mantido a 25 °C por 6 h. A autoclave foi ventilada e a mistura da reação foi removida. Permitiu-se que o dióxido de enxofre evaporasse enquanto o volume da solução era mantido constante pela adição lenta de éter seco. O cloridrato de cloro amorfo (11) foi filtrado, lavado com éter e seco no vácuo, MP: 130-135 °C dec. Rendimento: 3,5 g. O uso do álcool 9-beta-epimérico nessa reação produziu o mesmo cloreto com rendimento comparável.
4-Acetil-9-ciano-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (12)
Cianeto de sódio seco e em pó, 40 g, foi adicionado a 300 mL de cianeto de hidrogênio líquido gelado. A mistura foi agitada e resfriada em gelo, e 7,5 g do cloridrato amorfo bruto de 4-acetil-9-cloro-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo [4,3f/g]-quinolina (11) acima foram adicionados. A agitação foi continuada em 500 mL de RBF por 30 minutos, após o que o cianeto de hidrogênio foi rapidamente destilado sob pressão reduzida abaixo de aproximadamente 10 °C. O resíduo foi misturado com clorofórmio e água gelada, e a mistura resultante foi filtrada. A camada orgânica foi separada, e a fase aquosa foi extraída duas vezes com clorofórmio. Os extratos combinados foram secos com sulfato de magnésio, descoloridos e o solvente foi destilado no vácuo. O produto (12) foi cristalizado a partir de acetato de etila; rendimento de 3,3 g (54% do total com base no cloridrato de álcool), m.p. 172-174 °C. A recristalização com o mesmo solvente aumentou o p.m. para 181-182 °C.
Cianeto de sódio seco e em pó, 40 g, foi adicionado a 300 mL de cianeto de hidrogênio líquido gelado. A mistura foi agitada e resfriada em gelo, e 7,5 g do cloridrato amorfo bruto de 4-acetil-9-cloro-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo [4,3f/g]-quinolina (11) acima foram adicionados. A agitação foi continuada em 500 mL de RBF por 30 minutos, após o que o cianeto de hidrogênio foi rapidamente destilado sob pressão reduzida abaixo de aproximadamente 10 °C. O resíduo foi misturado com clorofórmio e água gelada, e a mistura resultante foi filtrada. A camada orgânica foi separada, e a fase aquosa foi extraída duas vezes com clorofórmio. Os extratos combinados foram secos com sulfato de magnésio, descoloridos e o solvente foi destilado no vácuo. O produto (12) foi cristalizado a partir de acetato de etila; rendimento de 3,3 g (54% do total com base no cloridrato de álcool), m.p. 172-174 °C. A recristalização com o mesmo solvente aumentou o p.m. para 181-182 °C.
9-Carbometoxi-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (13)
O produto (12) acima, 1,0 g, foi misturado com 15 mL de metanol e 0,25 mL de água. A mistura foi resfriada e 2 mL de ácido sulfúrico concentrado foram adicionados lentamente. A solução foi selada em um tubo de vidro sob nitrogênio e aquecida a 100 °C por 23 a 24 h em RBF de 100 mL com condensador de refluxo. A mistura foi tratada com carbono descolorido e, em seguida, concentrada no vácuo para cerca de 10 mL. Ela foi despejada em uma mistura de clorofórmio (30 mL), gelo e 10 g de bicarbonato de sódio. A camada de clorofórmio foi separada, e a fase aquosa foi extraída com 3 porções de 10 mL de clorofórmio. Os extratos combinados foram secos sobre sulfato de magnésio, evaporados até a secura e o produto (13) foi cristalizado a partir de benzeno; rendimento de 0,51 g (53%), MP: 159-160 °C. Foi recristalizado a partir de acetato de etila; MP: 160-161 °C.
O produto (12) acima, 1,0 g, foi misturado com 15 mL de metanol e 0,25 mL de água. A mistura foi resfriada e 2 mL de ácido sulfúrico concentrado foram adicionados lentamente. A solução foi selada em um tubo de vidro sob nitrogênio e aquecida a 100 °C por 23 a 24 h em RBF de 100 mL com condensador de refluxo. A mistura foi tratada com carbono descolorido e, em seguida, concentrada no vácuo para cerca de 10 mL. Ela foi despejada em uma mistura de clorofórmio (30 mL), gelo e 10 g de bicarbonato de sódio. A camada de clorofórmio foi separada, e a fase aquosa foi extraída com 3 porções de 10 mL de clorofórmio. Os extratos combinados foram secos sobre sulfato de magnésio, evaporados até a secura e o produto (13) foi cristalizado a partir de benzeno; rendimento de 0,51 g (53%), MP: 159-160 °C. Foi recristalizado a partir de acetato de etila; MP: 160-161 °C.
Ácido dl-lisérgico sintético (14)
Uma mistura de 9-carbometoxi-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (13), 3,9 g, e 78 mL de solução de hidróxido de potássio a 1,5% foi refluxada por 30 minutos sob nitrogênio. Adicionou-se arseniato de sódio hidratado, 8,5 g, e níquel Raney (16 g, úmido), previamente desativado por ebulição em suspensão de xileno, e a mistura foi aquecida sob refluxo e agitada em atmosfera de nitrogênio por 20 horas em RBF de 200 mL com condensador de refluxo. A solução foi tratada com carbono, e o ácido lisérgico bruto (14) foi precipitado por neutralização até pH 5,6. Ele foi filtrado e lavado com água; rendimento de 1,04 g, MP: 240-242 °C dec. Uma segunda safra, 0,16 g, MP: 233-235 °C dec. também foi obtida; rendimento total de 30%. O ácido pode ser purificado dissolvendo-o em hidróxido de amônio diluído, tratando-o com carbono descolorante e reprecipitando-o com dióxido de carbono, MP: 242-243 °C dec; uma mistura com ácido dl-lisérgico feita a partir de ácido d-lisérgico natural também foi de 242-243 °C dec. O ácido anidro foi obtido por secagem em vácuo por várias horas a 150 °C.
Uma mistura de 9-carbometoxi-7-metil-4,5,5a,6,6a,7,8,9-octa-hidroindolo-[4,3-fg]-quinolina (13), 3,9 g, e 78 mL de solução de hidróxido de potássio a 1,5% foi refluxada por 30 minutos sob nitrogênio. Adicionou-se arseniato de sódio hidratado, 8,5 g, e níquel Raney (16 g, úmido), previamente desativado por ebulição em suspensão de xileno, e a mistura foi aquecida sob refluxo e agitada em atmosfera de nitrogênio por 20 horas em RBF de 200 mL com condensador de refluxo. A solução foi tratada com carbono, e o ácido lisérgico bruto (14) foi precipitado por neutralização até pH 5,6. Ele foi filtrado e lavado com água; rendimento de 1,04 g, MP: 240-242 °C dec. Uma segunda safra, 0,16 g, MP: 233-235 °C dec. também foi obtida; rendimento total de 30%. O ácido pode ser purificado dissolvendo-o em hidróxido de amônio diluído, tratando-o com carbono descolorante e reprecipitando-o com dióxido de carbono, MP: 242-243 °C dec; uma mistura com ácido dl-lisérgico feita a partir de ácido d-lisérgico natural também foi de 242-243 °C dec. O ácido anidro foi obtido por secagem em vácuo por várias horas a 150 °C.
Attachments
Last edited: