
Killar jag har en fråga:
Finns det ett sätt att få 3,4-Methylenedioxyphenylpropan-2-one (MDP2P) från Eugenol?
Om ja kan någon ge mig en exakt syntes?
Jag hittade den här syntesen men jag förstår inte hur jag kan göra beta-nitroisoeugenol och β-nitroisosafrol.
Problemet är att det i mitt land är omöjligt att få sassafrasolja, safrol, isosafrol och piperonal.
I en 5-liters 3-halsad kolv utrustad med en omrörare och återflödeskondensor placerades 1000 ml etanol och 110 g beta-nitroisoeugenol. Blandningen upphettades under omrörning och när nitroisoeugenolen var löst tillsattes 2500 mL hett vatten. Under uppvärmning och kraftig omrörning tillsattes 200 g reducerat järnpulver och 8 g hydratiserad järn(III)klorid. Under fortsatt omrörning tillsattes långsamt 100 mL koncentrerad saltsyra. Saltsyran orsakade en våldsam reaktion som avtog efter ca 5 minuter; blandningen återflödades under omrörning i två timmar och destillerades sedan under reducerat tryck tills ca 2 liter destillat samlats upp. Återstoden filtrerades och den fluffiga järnoxiden tvättades noggrant med varmt vatten och sedan med eter. Det kombinerade filtratet och tvättvattnet surgjordes kraftigt med saltsyra och extraherades med eter. Etern torkades och destillerades för att ge en lätt olja som fraktionerades i vakuum för att ge 68 g vanillylmetylketon som kokade vid 126-127°C vid 0,3 mm (72% utbyte).
3,4-dimetoxifenylaceton och 3,4-metylendioxifenylaceton framställdes från motsvarande β-nitropropenylbensener genom nästan identiska förfaranden med 90 respektive 72 % utbyte.
För beredning av 2-MeO-P2P med toluen/vatten som lösningsmedel, se Organic Syntheses, Coll. vol. 4, s. 573.
Min modifiering av denna beredning:
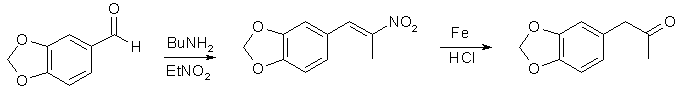
I en 2-liters 3-halskolv försedd med omrörare och återloppskylare placerades 460 ml etanol1 och 67 g β-nitroisosafrol2. Blandningen upphettades under omrörning och när de gula kristallerna var upplösta tillsattes 1100 ml hett vatten. Under uppvärmning och kraftig omrörning tillsattes 80 g reducerat järnpulver3 och 5 g FeCl3-6H2O. Under fortsatt omrörning tillsattes 63 ml konc. HCl på 30 min. Blandningen återflödade under omrörning i 2 timmar och destillerades sedan under atmosfärstryck4. Återstoden filtrerades och den bruna järnoxiden extraherades med 3x50 ml CH2Cl2. Filtratet och de organiska sköljningarna surgjordes med HCl och det röda ketonskiktet samlades upp. Ljusgrönt vattenskikt extraherades 2x100 ml CH2Cl2. De kombinerade extrakten torkades med Na2SO4 och destillerades vid atmosfärstryck (80°C bad). Diklormetan avlägsnades fullständigt i vakuum för att ge 55 g rå MDP2P som djupröd olja som användes för CH3NH2 / Al-reaktion.
Anmärkningar:
Finns det ett sätt att få 3,4-Methylenedioxyphenylpropan-2-one (MDP2P) från Eugenol?
Om ja kan någon ge mig en exakt syntes?
Jag hittade den här syntesen men jag förstår inte hur jag kan göra beta-nitroisoeugenol och β-nitroisosafrol.
Problemet är att det i mitt land är omöjligt att få sassafrasolja, safrol, isosafrol och piperonal.
I en 5-liters 3-halsad kolv utrustad med en omrörare och återflödeskondensor placerades 1000 ml etanol och 110 g beta-nitroisoeugenol. Blandningen upphettades under omrörning och när nitroisoeugenolen var löst tillsattes 2500 mL hett vatten. Under uppvärmning och kraftig omrörning tillsattes 200 g reducerat järnpulver och 8 g hydratiserad järn(III)klorid. Under fortsatt omrörning tillsattes långsamt 100 mL koncentrerad saltsyra. Saltsyran orsakade en våldsam reaktion som avtog efter ca 5 minuter; blandningen återflödades under omrörning i två timmar och destillerades sedan under reducerat tryck tills ca 2 liter destillat samlats upp. Återstoden filtrerades och den fluffiga järnoxiden tvättades noggrant med varmt vatten och sedan med eter. Det kombinerade filtratet och tvättvattnet surgjordes kraftigt med saltsyra och extraherades med eter. Etern torkades och destillerades för att ge en lätt olja som fraktionerades i vakuum för att ge 68 g vanillylmetylketon som kokade vid 126-127°C vid 0,3 mm (72% utbyte).
3,4-dimetoxifenylaceton och 3,4-metylendioxifenylaceton framställdes från motsvarande β-nitropropenylbensener genom nästan identiska förfaranden med 90 respektive 72 % utbyte.
För beredning av 2-MeO-P2P med toluen/vatten som lösningsmedel, se Organic Syntheses, Coll. vol. 4, s. 573.
Min modifiering av denna beredning:
I en 2-liters 3-halskolv försedd med omrörare och återloppskylare placerades 460 ml etanol1 och 67 g β-nitroisosafrol2. Blandningen upphettades under omrörning och när de gula kristallerna var upplösta tillsattes 1100 ml hett vatten. Under uppvärmning och kraftig omrörning tillsattes 80 g reducerat järnpulver3 och 5 g FeCl3-6H2O. Under fortsatt omrörning tillsattes 63 ml konc. HCl på 30 min. Blandningen återflödade under omrörning i 2 timmar och destillerades sedan under atmosfärstryck4. Återstoden filtrerades och den bruna järnoxiden extraherades med 3x50 ml CH2Cl2. Filtratet och de organiska sköljningarna surgjordes med HCl och det röda ketonskiktet samlades upp. Ljusgrönt vattenskikt extraherades 2x100 ml CH2Cl2. De kombinerade extrakten torkades med Na2SO4 och destillerades vid atmosfärstryck (80°C bad). Diklormetan avlägsnades fullständigt i vakuum för att ge 55 g rå MDP2P som djupröd olja som användes för CH3NH2 / Al-reaktion.
Anmärkningar:
- Användning av metanol istället för etanol ledde till ofullständig upplösning av gula kristaller.
- β-Nitroisosafrol kan enkelt framställas med Knoevenagel: 150 g piperonal blandades med 76 ml nitroetan (piperonal ska lösas upp helt för att ge färglös vätska) och 1,5 ml n-butylamin(eller n-pentylamin) tillsattes. Den resulterande gula lösningen förvarades på en mörk plats; lite vatten började dyka upp efter 2-3 dagar när reaktionen fortskred. Ljusorange kristaller av produkten började fällas ut efter 6-8 dagar. Efter 20 dagar filtrerades en blandning. Det ljusbruna filtratet filtrerades igen efter 5 dagar för att ge nästa skörd av produkten. Utbyte 85-90% av ljusorange kristaller, som är lämpliga för beredning av ketonen.
- Ett järn reducerat med väte användes. Användning av 20 mesh järnpulver istället för reducerat järn ledde till mindre kraftfull reaktion, resulterande etanol innehöll inte NH3 och keton förorenades med cirka 20% av dess oxim (inte kvantitativ reduktion).
- En fraktion 77-85°C samlades in. Den innehöll lite NH3 som kan neutraliseras med konc. H2SO4 och destilleras igen via Vigreux-kolonnen. Resulterande 90% etanol användes för beredning av nästa del av keton.
